 Open Access Article
Open Access ArticleLarge-scale synthesis of nanocrystals in a multichannel droplet reactor†
Adrian M.
Nightingale
a,
James H.
Bannock
a,
Siva H.
Krishnadasan
a,
Flannan T. F.
O'Mahony
a,
Saif A.
Haque
a,
Jeremy
Sloan
b,
Chris
Drury
c,
Robert
McIntyre
c and
John C.
deMello
*a
aCentre for Plastic Electronics, Department of Chemistry, Imperial College London, Exhibition Road, South Kensington, London SW7 2AY, UK. E-mail: j.demello@imperial.ac.uk
bDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK
cMillennium Inorganic Chemicals, Laporte Road, Stallingborough, Lincolnshire DN40 2PR, UK
First published on 5th February 2013
Abstract
We report a multichannel microfluidic droplet reactor for the large-scale, high temperature synthesis of nanocrystals. The reactor was applied here to the production of CdTe, CdSe and alloyed CdSeTe nanocrystals, and found in all cases to provide high quality quantum dots with spectral properties that did not vary between channels or over time. One hour test runs yielded 3.7, 1.5 and 2.1 g of purified CdTe, CdSe and the alloy, respectively, using 0.4 M cadmium precursor solutions and carrier and reagent phase flow rates of 4 and 2 ml min−1. A further nine hour test-run applied to CdTe, utilizing increased carrier and reagent flow rates of 5 and 3 ml min−1, yielded 54.4 g of dry purified material, corresponding to a production rate of 145 g per day. The reactor architecture is inherently scalable and, with only minimal modifications, should allow for straightforward expansion to the kilogram-per-day production levels sought by industry.
Introduction
Nanocrystals provide significant advantages over bulk materials in terms of enhanced processability, chemical activity, bioavailability, and magneto and optoelectronic functionality.1 However to exploit such properties requires exacting control over size, shape and chemical composition. Effective strategies for achieving such control have been developed for gram-scale batch syntheses,2 but raising production levels to the larger kilogram quantities needed for many commercial applications has proven to be difficult. Increased reaction volumes typically lead to diminished mixing efficiencies and poorer heat management, causing spatial variations in temperature and chemical composition that influence localised reaction kinetics and induce polydispersity in the final product or lead to unwanted side reactions, lowering yields.In 2002, as an alternative to batch synthesis, Edel et al.3 proposed the use of microscale flow reactors for nanocrystal synthesis, reasoning that the smaller reaction volumes would provide greater homogeneity in the chemical environment, thereby improving product quality. In subsequent years microreactors have been extensively applied to the small-scale synthesis of high quality nanoparticles.4 Nanocrystalline colloids of a wide range of materials have been prepared in microreactors including (but not limited to) CdSe,5 CdS,3,6 InP,7 Ag,8 Au,9 Co,10 TiO2,11 SiO2,12 LaPO4,13 CaCO3,14 FexOy,15,16 zeolites17,18 and BaSO4,19 as well as more complicated core/shell structures such as CdSe/ZnS,20 Fe2O3/SiO2,21 SiO2/Au,22 Au/Ag/Au23 and CdSe/ZnSe/ZnS.24 In all cases, microreactors were shown to offer significant advantages over equivalent batch reactions, most notably in terms of increased reaction control and the ability to fine tune the physicochemical properties of the final product. However microfluidic methods have until now been limited to the small-scale synthesis of nanomaterials (≲1 g per day), and their applicability to high volume materials production has remained untested.
The throughput of microfluidic reactors may be raised through a combination of increased reagent concentrations, higher flow rates and scaling-out, i.e. operating N identical reaction channels in parallel to achieve an N-fold increase in throughput relative to a single-channel reactor. Scaling out is an especially attractive approach since it requires no changes to the reactor geometry, and so allows materials throughput to be increased without any modifications to the underlying process chemistry. Hence, in contrast to the scaling up of batch reactions, scaling out can in principle be implemented without any detriment to product quality or yield.
Despite its obvious importance, scaling out has been a largely neglected aspect of microreactor technology. There are only a handful of reports of its successful application to chemical syntheses (nanoscale or otherwise), most of which have used gas/liquid reactors.25–27 One of the most notable examples of scale-out was reported by Chambers and co-workers, who fabricated a multi-channel F2 (g)/liquid based flow reactor for the direct fluorination of (liquid) organic molecules with an output of ∼150 g per day.25 Comparable-throughput micro-reactors for nanocrystal synthesis would easily satisfy current industrial demands for quantum dots and many other low-volume/high-value nano-crystals, provided adequate materials quality could be assured.
The principal challenge for achieving successful scale-out is the development of a scalable architecture that allows multiple channels to be supplied from a small number of communal feed reservoirs,‡ while ensuring excellent consistency of product from channel to channel and over time. Crucially, individual channels must exhibit excellent operating stabilities and remain fully operational over extended periods of time. If the channel number N is sufficiently large and all channels are identical with a common mean-time-before-failure (MTBF) τf, the number of working channels will decay exponentially with a lifetime τf (or faster if failures are positively correlated); hence, after a time t = τf, almost a third of the channels will already be defective, leading to an appreciable drop in yield and/or significant product inhomogeneity (depending on the nature of the failure mode and reaction chemistry).28 Achieving a high MTBF is therefore essential for reliable scale-out.
Most reports of nanoparticle synthesis in microreactors have involved single phase reactors that manipulate miscible streams of a single reagent phase. Such reactors can be problematic for scale-out synthesis since they are susceptible to fouling under prolonged operation – a consequence of the reagent phase making physical contact with the channel walls.29 By selecting an immiscible carrier fluid that preferentially wets the walls and injecting it into the channel alongside the reagent phase, the latter can be forced to separate into a train of discrete droplets that are completely encapsulated within the carrier fluid. Importantly the droplets are swept through the channel at a common speed (ensuring excellent uniformity of flow) and remain fully isolated from the walls, greatly reducing the risk of reactor fouling.30 Droplet flow is therefore well suited to applications where high reliability is required from multiple channels, and might therefore be expected to provide a sufficiently high MTBF for successful scale-out.
Droplet-based synthesis of nanocrystals was first reported in 2004 by Shestopalov et al., who carried out an aqueous-based synthesis of CdS quantum dots using a single channel water-in-fluorous-oil droplet reactor fabricated from poly(dimethylsiloxane).31 They reported excellent stability of the reactor when operating in droplet mode, and observed rapid fouling of the reactor when the fluorous phase was replaced with water and the reactor reverted to a single-phase mode of operation. Single channel droplet reactors have subsequently been applied to the low temperature (≲100 °C) synthesis of many nanocrystalline materials, with good resilience to fouling being reported in virtually all cases.14,16,18,23
In practice, low temperature synthesis routes tend to yield crystals with rather high defect densities and for optimal crystalline quality high temperature routes must be used to anneal out crystal defects.32 To our knowledge there have been just three reports of nanocrystal synthesis in droplet reactors operating substantially above 100 °C17,33,34 due to the difficulty of maintaining reliable droplet flow under strong heating.§ High temperature nanocrystal synthesis in a droplet reactor was first reported by Chan et al.33 who prepared CdSe quantum dots in octadecene (ODE) droplets, using an immiscible high boiling-point perfluorinated polyether (PFPE) carrier fluid. The reaction was carried out at temperatures of up to 300 °C in a glass microfluidic chip which had been surface-modified with a perfluoroalkylsilane to ensure preferential wetting of the channel walls by the fluorous carrier phase. High quality CdSe was obtained during the first five hours of operation, with controlled droplet flow and no fouling of the reactor, confirming the effectiveness of the droplet approach. However continued operation resulted in degradation of the silane surface-coating, causing the reagent phase to wet the channel walls and droplet flow to cease.
Following Chen et al. we subsequently reported high temperature droplet synthesis of CdSe in the same ODE–PFPE fluid system, but using a polytetrafluoroethylene (PTFE) capillary instead of a glass microfluidic chip – PTFE being chosen as it is preferentially wetted by the fluorous carrier phase without the need for any surface treatment.34 The capillary-based droplet reactor could be operated indefinitely at temperatures up to 250 °C, yielding particles with well-defined band-edge emission spectra that did not vary over the course of a day's production. A similar PTFE capillary-based reactor has since been successfully applied by Hoang et al. to the high temperature synthesis of zeolites.17
Owing to the excellent stability of our earlier single-channel PTFE capillary reactor,34 we sought to develop a multichannel scale-out reactor for high temperature nanocrystal synthesis using a similar capillary-based architecture. Our goal was to develop a fully functional five-channel prototype droplet reactor capable of producing high quality quantum dots at rates in excess of one hundred grams per day, using a scalable architecture that would allow for straightforward expansion to the kilogram-per-day production levels sought by industry. The fabrication of the scale-out reactor and its application to the volume synthesis of cadmium chalcogenide nanocrystals is described below.
Reactor design
Fig. 1 shows a schematic of the scale-out reactor. For maximum scalability, all channels of the reactor were supplied from a common set of feed reservoirs. For the reagent phase, a premixed precursor solution of chalcogen–trioctylphosphine and cadmium oleate in octadecene (see Experimental) was loaded into a single two-litre storage bottle. Samples of the precursor solution were continuously extracted from the bottle into two 5 ml syringe pumps (Harvard Pump 11+) connected in a push/pull arrangement using a pair of two-way solenoid valves (Bio-chem valve, 100T3MP24-62, controlled using a KMtronic USB relay board).¶ The pumps and valves were programmatically controlled so that one syringe would withdraw fresh precursor solution from the bottle as the other pulselessly dispensed its payload into the reactor, with the two syringes changing direction and swapping roles when the first syringe was almost full and the second almost empty.|| In a production environment continuous pumps – HPLC or gear – would be more appropriate for delivery of the reagent phase, but syringe pumps were the preferred option here for safely handling large quantities of cadmium compounds since they could be readily dismantled and cleaned. | ||
| Fig. 1 Schematic of multichannel droplet reactor. | ||
For the carrier phase a small plastic beaker with an overflow spout was filled with perfluorinated polyether (Fomblin Y 06/6) up to the level of the overflow (see Fig. 1). This beaker was used both for storage of the carrier fluid and for receipt of the effluent at the reactor outlet as described below. One end of a PTFE capillary was immersed in the PFPE and used to continuously withdraw carrier fluid into the inlet of a high pressure liquid chromatography (HPLC) pump (Laballiance 1500), selected for its ability to maintain a continuous flow of (viscous) PFPE into the rest of the reactor against a high back-pressure.
The outlets of the two pumping systems were separately fed into five-way passive flow-dividers, which split the precursor and the carrier fluid into parallel streams. Paired streams of precursor and carrier fluid were then individually merged at five separate PTFE T-junctions (see Experimental). The outlets of the T-junctions were connected to 5 m PTFE reaction channels (inner diameter = 1 mm), yielding five parallel streams of reagent-phase droplets in PFPE carrier fluid. Carrier to reagent flow-rate ratios of at least 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 were used to ensure a sufficient excess of carrier fluid to fully wet the channel walls.
:2 were used to ensure a sufficient excess of carrier fluid to fully wet the channel walls.
To ensure droplets in each line experienced equivalent temperature profiles – essential for ensuring consistency of product – the five reaction channels were intertwined into a single cord using a symmetric braid: a five-stranded English sinnet35 (see Fig. 1 and 7b). A 2.65 m inner section of the braided cord was immersed in a heated oil bath to induce nucleation and growth of the nanocrystals. As a result of the braiding all droplets moved helically around the central axis of the cord, averaging out the effects of temperature gradients and inhomogeneities.
The effluent from each line was delivered into the same beaker that was used for storage of the PFPE carrier fluid, with the reaction phase forming a discrete layer above the PFPE (which was recycled into the main reactor by the HPLC pump, thereby avoiding the cost and inconvenience of continuously supplying fresh carrier fluid). The level of the reaction phase gradually increased due to the continuous withdrawal of precursor solution from the two-litre bottle, while the level of the (recirculating) carrier fluid remained constant. In consequence the upper surface of the reaction phase periodically rose above the level of the overflow, causing a slug of solution to be expelled from the beaker into a secondary collection vessel. (Note, this process was periodic rather than continuous since a sufficient body of fluid had to first accumulate in the beaker for the pressure head to overcome surface tension at the exit of the overflow spout and so allow the excess reagent phase to separate from the remainder of the solution.)
Ensuring balanced flow through the different reactor lines is of critical importance for attaining an homogenous product, and is recognised as being one of the principal obstacles to achieving successful scale-out.36 Passive flow dividers of the kind used here partition a fluid stream on the basis of pressure, and slight differences downstream (due e.g. to diameter variations, kinks and other physical imperfections in the lines) can therefore result in unbalanced flow.
To achieve balanced carrier flow, the following equalisation procedure was employed. The reagent flow was first set to zero and carrier fluid was pumped into the flow divider at a steady rate of 3 ml min−1 from the HPLC pump, corresponding to an average throughput per line of 0.6 ml min−1. Samples from each line were collected over a two minute period and weighed on a balance. Significant variations in carrier flow rate (Qc) were initially observed across the lines, with lines 2, 3 and 4 operating at ∼1.03 g min−1 and lines 1 and 5 at ∼1.16 g min−1. To balance the flow rates, the back-pressures of the slow-flowing lines were reduced by trimming their free (unbraided) ends by equal amounts until the back-pressures were equalised and even flow attained.**Fig. 2a shows the spread in Qc values after reducing the length of the slow-flowing lines by 0, 20, 40 and 60 cm. The relative standard deviation (RSD = standard deviation divided by the mean) decreased rapidly as the slow lines were shortened, falling from an initial value of 6.9% to a final value of 0.4% after trimming 60 cm.
 | ||
| Fig. 2 (a) Carrier flow rates Qc in each of the five reactor lines versus length of tubing removed from (slow-flowing) lines 2, 3 and 4. (b) Total flow-rate QT (= carrier flow rate Qc plus reagent phase flow rate QR) versus calculated back pressure for each of the five reactor lines, using pressure-drop channels of varying inner diameter (ID) and length (l). (c) Total flow-rate QT in each of the five reactor lines versus time over a one hour CdTe production run. The horizontal dotted lines in each plot denote percentage deviations from the mean flow rate. | ||
With the carrier flow balanced, ODE was then introduced at a flow rate of 0.6 ml min−1 (0.47 g min−1), while maintaining the carrier flow at 3.0 ml min−1 (5.4 g min−1). This resulted in a substantial deterioration in the line-to-line flow uniformity from RSD = 0.4% without the reagent phase to RSD = 9.4% with the reagent phase. (Note, here the RSDs refer to the total flow-rate QT, i.e. the flow-rate of the carrier fluid Qcplus the flow-rate of the reagent phase QR.) To restore the flow uniformity, pressure-drop channels (PDCs) – identical short lengths of narrow diameter tubing – were inserted between the outlets of the reagent-phase flow divider and the T-junctions. PDCs provide a high back-pressure immediately before the main reactor lines and have previously been reported to provide improved flow uniformity by reducing the relative influence of pressure variations and fluctuations downstream.27,37
PDC tubing of different lengths and inner diameters was evaluated with a view to attaining well balanced flow without incurring excessive back-pressures that might place undue strain on the syringe pumps. Fig. 2b shows for PDCs of various lengths and diameters the total measured flow-rate through each line versus the calculated back-pressure Δp, determined according to the Hagen–Poiseuille formula, which for cylindrical channels of length l, diameter d and constant flow rate is given by: Δp ∝ l/d4. (Note, for simplicity we assumed a common friction factor and normalised all back-pressures to the d = 800 μm, l = 300 mm case.) The spread in QT values was seen to diminish rapidly with increasing back-pressure, decreasing to an RSD of 0.6% when using 150 μm fused silica capillaries of length 300 mm. These capillaries were accordingly selected for the pressure drop channels. In this way, by first adjusting the carrier flow via back-pressure adjustments and then using PDCs to regulate the reagent phase, a stable balanced flow distribution was obtained.
Results and discussion
The scale-out reactor was first applied to the synthesis of CdTe nanocrystals, using a precursor of pre-mixed cadmium oleate and tellurium trioctylphosphine (TeTOP) in octadecene (2:1 molar ratio, see Experimental). The oil bath temperature was 175 °C and the precursor and carrier flow-rates were set to 4 and 2 ml min−1, respectively implying a reaction time of 1.7 minutes for the selected flow-rates. The time and temperature were selected on the basis of exploratory experiments carried out using a single-line droplet reactor34 with the same precursor solution.
In operation, each T-junction expelled a regular train of discrete reagent-phase droplets into a separate line of the reactor. On entering the braided zone the droplets adopted helical trajectories, with droplets from different lines interweaving amongst one another in accordance with the sinnet motif. On entering the oil-bath all droplets had a uniform size, speed and separation but the uniformity was gradually disrupted as they passed through the heated zone due to occasional thermocoalescence of adjacent droplets. (Note, coalescence did not have a noticeable effect on residence times and hence did not affect the final particle properties – see results below.) The colour of the droplets progressively darkened as they passed through the oil bath from a pale yellow at the entry point to a deep red at the exit point, consistent with the formation of CdTe nanocrystals.
In normal operation, the effluent from all five lines was collected in the same plastic beaker used for phase separation and PFPE storage, with the reagent phase periodically ‘spilling’ over into a secondary collection vessel as described above. In addition for analysis purposes 2 min aliquots were collected directly from each line at ten minute intervals over the course of one hour and stored in separate vials (thirty five samples in total). The flow-rates determined from the aliquot masses were consistent from line-to-line and over time as shown in Fig. 2c.
Fig. 3a shows emission spectra for the samples collected from each line over the course of an hour (see ESI Fig. S1† for corresponding absorption spectra); no significant differences in spectra were observable from line to line or over time, indicating well balanced and stable (drift-free) operation of the reactor. The excellent consistency of product between lines is clear from Fig. 3b, in which absorption and emission spectra for samples collected from all five lines at time t = 40 min are shown in a single plot. The close agreement between spectra is consistent with the production of nanocrystalline CdTe of a common size, shape and chemical composition.
 | ||
| Fig. 3 (a) Normalised solution-phase emission spectra of CdTe collected from each of the five reaction lines at ten minute intervals over a period of one hour. (b) Normalised solution-phase absorption spectra (dashed lines) and emission spectra (solid lines) of CdTe collected from each of the five reaction lines at t = 40 min. (c and d) Time-dependence of peak emission wavelength (λmax) and full width half maxima (FWHM) of CdSe, CdTe and alloyed CdSeTe nanocrystals collected from each of the five reaction lines during one hour production runs. | ||
The reactor was further applied to the synthesis of CdSe and alloyed CdSeTe – the latter material being of interest for its low optical gap.38,39 The CdSe precursor comprised a 2:1 molar mixture of cadmium oleate with SeTOP, while the alloy precursor comprised a 10:4:1 molar blend of cadmium oleate with SeTOP and TeTOP (see Experimental). In both cases the total reagent concentration, the cadmium to chalcogenide ratio, and the ligand concentration matched those used previously for CdTe. The temperature of the oil-bath was set to 175 °C for CdSe and to 195 °C for the alloy, with all other experimental parameters (flow rates, tubing lengths) being kept the same as before. Visually, the droplet flow was in both cases similar to that observed for CdTe, with droplets undergoing occasional thermocoalescence as they passed through the oil-bath and changing in colour from pale yellow to orange in the case of CdSe and from orange to deep red/brown in the case of the alloy.
2 min aliquots were again collected from each line at ten minute intervals over the course of one hour. As with CdTe, the flow rates were well balanced (see ESI Fig. S2†), and there were no significant differences in spectra between lines or over time (see ESI Fig. S3 and S4†). Fig. 3c and d show for each material how the peak wavelengths (λmax) and full width half maxima (FWHM) of the emission spectra varied over the course of the one hour production runs. λmax for each material fluctuated by only a few nanometers (standard deviations of 0.9, 0.8 and 2.7 nm) about the mean values of 596, 525 and 678 nm for CdTe, CdSe and the alloy respectively, providing further evidence of the uniform, stable nature of the reactor. The longer wavelength emission from the alloy is consistent with the occurrence of band-gap bowing in which (for a given particle size) the optical gap of the daughter material dips below that of the two parent materials with a parabolic dependence on composition.38,39
In normal operation (i.e. when not sampling aliquots from individual lines), the combined effluent from the five channels accumulated in the collection vessel, see Fig. 1. Photographs of CdTe, CdSe and alloy solutions taken from the collection vessel are shown in Fig. 4a under conditions of UV excitation, with the corresponding emission spectra shown in Fig. 4b. As expected the spectra of the combined samples matched closely to those obtained from the individual reactor lines, with λmax values of 596, 525 and 674 nm and FWHM values of 43, 34 and 90 for CdTe, CdSe and the alloy respectively.
 | ||
| Fig. 4 (a) Photograph of reactor-synthesised CdSe (left), CdTe (middle) and alloy (right) under UV illumination; samples were taken from the collection vessel (see Fig. 1), and so contained a balanced mixture of effluent from all five reaction lines. (b) Photoluminescence spectra for the same three samples under 360, 390 and 420 nm excitation respectively. | ||
Each material was purified using standard precipitation/centrifugation protocols and dried overnight in a vacuum oven at 50 °C (see Experimental). Hour long runs produced 3.7, 1.5 and 2.1 g of purified CdTe, CdSe and the alloy respectively. The relative yields reflect the greater molar mass of CdTe over CdSe and the higher relative reactivity of TeTOP over SeTOP.
The three materials were characterized using transmission electron microscopy (TEM), X-ray diffraction (XRD) and energy dispersive X-ray (EDX) analysis. TEM showed the purified materials to be weakly anisotropic branched nanostructures with smallest dimension <3 nm (see Fig. 5a–d). The observed morphology is consistent with anisotropic crystal growth driven by the high reagent concentrations and the use of a bulky chalcogen ligand (TOP).40 High resolution TEM images revealed the crystalline nature of the nanostructures, with lattice fringes clearly resolvable for all three materials (see Fig. 5d and ESI Fig. S5†). [1 1 1] d-spacings were identifiable in all three cases and determined via a Fourier transform of the images to be 3.4, 3.87 and 3.56 Å for CdSe, CdTe and the alloy respectively, in broad agreement with literature values.41 The intermediate d-spacing in the latter case indicates the successful formation of an alloyed structure, which was further confirmed by XRD.
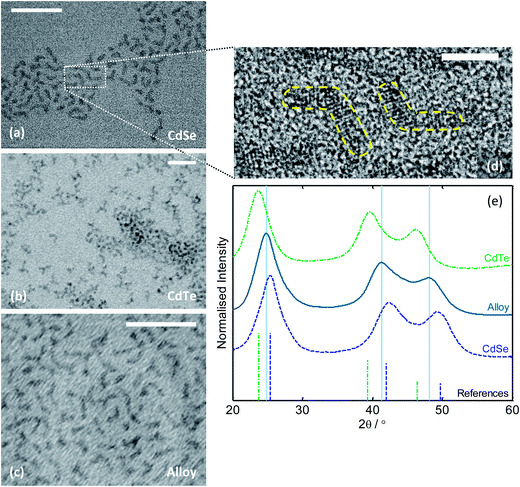 | ||
| Fig. 5 (a–c) Transmission electron micrographs of reactor-synthesised CdSe, CdTe and alloyed CdSeTe; scale-bars equal 30 nm. (d) High resolution TEM image of CdSe, showing “boomerang”-shaped nanocrystals with resolvable lattice fringes; scale-bar equals 5 nm. (e) XRD spectra of the three materials, with reference lines shown for zinc blende CdSe (blue) and zinc blende CdTe (green). | ||
As shown in Fig. 5e, the XRD spectra for the three materials matched closely in terms of shape and line-width, although the peak locations were somewhat shifted – consistent with their having a common crystal structure but different lattice parameters. The CdSe and CdTe peaks correlated well with the standard zinc blende reference peaks (JCPDS cards 00-019-0191 and 00-015-0770 respectively), while the alloy showed features at locations intermediate between the two – consistent with the successful formation of an alloyed zinc blende crystal structure with an intermediately sized unit cell. Applying the Scherrer equation to the [1 0 0] peak of each material (at 2θ ≈ 25°) yielded in all cases a crystallite size of ∼2.5 nm, in broad agreement with the small dimension of the structures seen in the transmission electron micrographs. EDX showed the alloy to have a Se:Te ratio of ∼2:1 (see ESI Fig. S6†). The relative composition of the alloy nanocrystals differed substantially from the 4:1 composition of the precursor solution due to the higher reactivity of the Te precursor species.38,42
Finally, to evaluate the viability of applying the scale-out reactor to the sustained production of nanocrystals at even higher levels of throughput, a nine hour test run was carried out for CdTe, using increased carrier and reagent flow-rates of 5 and 3 ml min−1 (which corresponded to the upper capabilities of the pumps we employed). All other reaction parameters, including capillary lengths, were kept the same as before. 30 s aliquots were collected from each line at one hour intervals over the nine hour duration of the run, and absorption and emission spectra recorded. As before, the flow rates of the carrier and reagent phases were well balanced and there were no significant differences in the measured spectra between lines or over time, as shown in Fig. 6 (see ESI Fig. S7† for the corresponding absorption spectra). The peak wavelength and full width half maxima of the emission spectra fluctuated about their mean values of 589.5 and 43.3 nm with small standard deviations of 1.0 and 0.6 nm respectively, confirming the good uniformity and stability of the reactor and the excellent consistency of the resultant product even at the elevated flow rates. Following purification 54.4 g of final dry material was recovered from the nine hour run, corresponding to a high production rate of 145 g per day. An image of the purified material is shown in Fig. 7a.
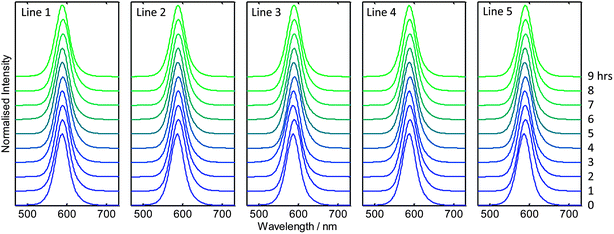 | ||
| Fig. 6 Normalised solution-phase emission spectra of CdTe collected from each of the five reaction lines at one hour intervals over a period of nine hours. | ||
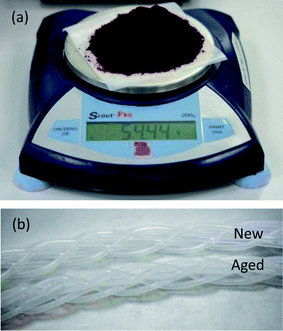 | ||
| Fig. 7 (a) Photograph of 54.4 g of purified CdTe from nine-hour production run. (b) Photograph of braided PTFE reaction lines before and after use; used lines showed no signs of fouling or aging. | ||
Fig. 7b shows a photograph of a section of the five braided reaction lines after extended use, together with a braid of unused PTFE capillary. No significant differences were observable between the pristine and used lines, with no visible evidence of particle deposition or reactor wear after use. Anecdotally we note that the same reactor lines were used for all measurements reported here and we have observed no degradation of the capillaries over several months of testing, indicating the effectiveness of the PFPE carrier fluid in protecting the channels against fouling and preserving the long-term operability of the reactor.
Conclusion
In conclusion we have developed a five-channel prototype droplet reactor for the large scale synthesis of nanocrystals at high temperature. The reactor was applied here to the production of nanocrystalline CdTe, CdSe and alloyed CdSeTe, and found in all cases to provide high quality quantum dots with spectral properties that did not vary between channels or over time. Initial one hour test runs yielded 3.7, 1.5 and 2.1 g of purified CdTe, CdSe and the alloy, respectively, using 0.4 M cadmium precursor solutions and carrier and reagent phase flow rates of 4 and 2 ml min−1. A further nine hour test-run applied to CdTe, utilizing increased carrier and reagent flow rates of 5 and 3 ml min−1, yielded 54.4 g of dry purified material, corresponding to a production rate of 145 g per day.The quantity of material produced here compares favourably with reports of high volume nanocrystal synthesis elsewhere in the literature where the term “large-scale” has been applied to the batch synthesis of 2.5 g of core/shell CdSe/CdS,43 2.7 g of InPZnS,44 2.8 g of ZnO,45 “several grams” of Fe3O4,46 9.6 g of CdSe,47 ≳10 g of Cu2ZnSnSe4,48 ≳10 g of CeO249 and ∼20 g of ZrO2;50 while the term “ultra large-scale” has been used to describe the batch synthesis of 40 g of Fe3O4.51 Although the reactor was operated here for a maximum of nine hours to avoid producing an excessive quantity of CdTe waste material, it showed no signs of aging during the course of the run, suggesting it should be amenable to many days of continuous operation. The 145 g per day level of throughput – whilst in itself a significant production rate for nanomaterials – was obtained using a modest five-way level of parallelisation. With only minimal changes to the reactor architecture, it should be quite feasible to expand the architecture to forty channels or more, providing ready access to production rates in excess of 1 kg per day.
Experimental
Materials
CdO (99.5%), Te (99.5%, 200 mesh powder), TOP (90%) and ODE (90%) were obtained from Alfa Aesar. Se (99.5%, 100 mesh powder) and oleic acid (90%) were obtained from Sigma Aldrich. Anhydrous ethanol and toluene were obtained from VWR. All chemicals were used as received.Cd oleate stock solution
To make 100 ml of Cd oleate stock solution, 5.4 g of CdO was added to 54.4 ml of oleic acid and 45.6 ml of ODE in a 250 ml round bottomed flask (giving a Cd:oleic acid molar ratio of ∼4 and a Cd concentration of 0.4 M). The resulting red/brown suspension was degassed for ten minutes under vacuum while heating to 100 °C. N2 was added in and the suspension heated to 180 °C with vigorous stirring. After 1 hour at this temperature the flask contents had cleared to give a clear solution of Cd oleate, with a very pale yellow colour. The flask was taken off the heat and allowed to cool.
TeTOP stock solution
To make 100 ml of TeTOP stock solution, 27.4 ml of TOP was first added to a 100 ml round bottomed flask under air-free conditions. 2.56 g of Te powder was added (giving a Te:TOP molar ratio of ∼3), the resulting suspension was degassed under vacuum for ∼15 minutes and then heated to ∼270 °C under N2 with vigorous stirring. Once the contents of the flask had turned from a turbid grey suspension to a clear yellow/green solution, the flask was removed from the heat and allowed to cool. After cooling to a clear orange colour, 72.6 ml of ODE was added to the solution giving a Te concentration of 0.2 M.
SeTOP stock solution
To make 100 ml of SeTOP stock solution, 27.4 ml of TOP and 72.6 ml of ODE were added to a 100 ml round bottomed flask under air-free conditions. 1.59 g of Se powder was added (giving a Se:TOP molar ratio of ∼3 and a Se concentration of 0.2 M) and left stirring until the Se had dissolved to give a clear colourless solution.
Nanocrystal precursor solutions
For the CdTe precursor solution, equal volumes of Cd oleate and TeTOP stock solutions were mixed to give a clear solution. The mixture was then left overnight before use. On initial mixing the solution was a bright yellow but darkened overnight to give a deep orange colour.The CdSe precursor solution was prepared in an identical fashion and was observed to change from colourless upon initial mixing to a light yellow colour after aging overnight.
For the alloy precursor solution, individual CdTe and CdSe precursor solutions were prepared as described above and then, after aging overnight, were combined (4:1 by volume CdSe:CdTe precursor solutions) and used immediately.
Characterisation
Low resolution TEM imaging was performed with a JEOL2010 microscope operated at 200 eV using formvar/carbon covered Cu grids (Agar). High resolution TEM imaging was performed with a 2100 LaB6 microscope fitted with a HR pole piece, operated at 200 kV, using 6 nm amorphous carbon coated 3.05 mm Cu grids (Agar).EDX was performed using an Oxford Instruments INCA 80 mm X-Max detector fitted to the JEOL2010 microscope. Three areas were sampled for each material in order to obtain an average atomic concentration.
Powder XRD was performed with a PANalytical X'Pert Pro MRD diffractometer, using Ni filtered Cu K-alpha radiation at 40 kV and 40 mA.
All photoluminescence and absorption measurements were carried out using a modified Jobin-Yvon Fluoromax 2 fluorimeter on unpurified reaction solution samples diluted using toluene (with the exception of Fig. 4b which shows the spectra of purified samples dissolved in toluene). A photodiode in the fluorimeter was used to monitor transmitted light through the cuvettes for simultaneous absorption measurements. For ease of comparison emission spectra were normalised with respect to the peak intensity, while absorption spectra were normalised with respect to the absorbance at the first excitonic peak.
Purification
The nanocrystals were purified using two precipitation–centrifugation–washing cycles. They were first precipitated from the reaction solution using anhydrous ethanol. Following isolation by centrifugation (5000 rpm for 20 minutes), the resulting nanocrystal pellet was washed with anhydrous ethanol, dried in a stream of nitrogen, and then re-dissolved in the minimum volume of toluene. The nanocrystals were once again precipitated with anhydrous ethanol, isolated by centrifugation (5000 rpm, 20 minutes), washed and then dried overnight in a vacuum oven at 50 °C.Fabrication of T-junctions
The T-junctions were fabricated in-house using a 4-dimensional CNC mill to machine PTFE round-stock (RS). As shown in ESI Fig. S8,† the T-junctions had 1 mm through-channels and were interfaced with the capillary tubing using flangeless ferrules and nuts (P-305, P-342, Upchurch Scientific).Acknowledgements
A. M. N. and S. H. K. thank the Engineering and Physical Sciences Research Council (EPSRC) for financial support through the Knowledge Transfer Secondment scheme. J. B. is funded under an EPSRC Doctoral Training Centre in Plastic Electronics (grant number EP/G037515/1) and holds an Industrial Fellowship with the Royal Commission for the Exhibition of 1851. J. S. is grateful to the Warwick Centre for Analytical Science (EPSRC funded grant EP/F034210/1). J. dM. is a Royal Society Industry Fellow. The authors also acknowledge financial support from the EPSRC under grant number EP/G031088/1.References
- U. Banin and O. Millo, in Nanoparticles, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 305–367 Search PubMed; E. Katz, A. N. Shipway and I. Willner, in Nanoparticles, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 368–421 Search PubMed; C. N. R. Rao, P. J. Thomas and G. U. Kulkarni, Nanocrystals: Synthesis, Properties and Applications; Springer, 2007; vol. 95 Search PubMed.
- T. Kim, S. W. Kim and M. Kang, J. Phys. Chem. Lett., 2012, 3, 214–218 CrossRef CAS; M. Protiere, N. Nerambourg, O. Renard and P. Reiss, Nanoscale Res. Lett., 2011, 6, 472 CrossRef.
- J. B. Edel, R. Fortt, J. C. deMello and A. J. deMello, Chem. Commun., 2002, 1136–1137 RSC.
- A. Abou-Hassan, O. Sandre and V. Cabuil, Angew. Chem., Int. Ed., 2010, 49, 6268–6286 CrossRef CAS; A. M. Nightingale and J. C. de Mello, J. Mater. Chem., 2010, 20, 8454–8463 RSC.
- E. M. Chan, R. A. Mathies and A. P. Alivisatos, Nano Lett., 2003, 3, 199–201 CrossRef CAS; H. Nakamura, Y. Yamaguchi, M. Miyazaki, H. Maeda, M. Uehara and P. Mulvaney, Chem. Commun., 2002, 2844–2845 RSC; B. K. H. Yen, N. E. Stott, K. F. Jensen and M. G. Bawendi, Adv. Mater., 2003, 15, 1858–1862 CrossRef.
- Z. Wan, H. Yang, W. Luan, S. T. Tu and X. Zhou, Nanoscale Res. Lett., 2009, 5, 130–137 CrossRef.
- J. Baek, P. M. Allen, M. G. Bawendi and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 627–630 CrossRef CAS; A. M. Nightingale and J. C. de Mello, ChemPhysChem, 2009, 10, 2612–2614 CrossRef.
- S. T. He, T. Kohira, M. Uehara, T. Kitamura, H. Nakamura, M. Miyazaki and H. Maeda, Chem. Lett., 2005, 34, 748–749 CrossRef CAS; J. M. Kohler, M. Held, U. Hubner and J. Wagner, Chem. Eng. Technol., 2007, 30, 347–354 CrossRef; Z. L. Xue, A. D. Terepka and Y. Hong, Nano Lett., 2004, 4, 2227–2232 CrossRef.
- D. Shalom, R. C. R. Wootton, R. F. Winkle, B. F. Cottam, R. Vilar, A. J. deMello and C. P. Wilde, Mater. Lett., 2007, 61, 1146–1150 CrossRef CAS; J. Wagner, T. Kirner, G. Mayer, J. Albert and J. M. Kohler, Chem. Eng. J., 2004, 101, 251–260 CrossRef; J. Wagner and J. M. Kohler, Nano Lett., 2005, 5, 685–691 CrossRef.
- Y. J. Song, L. L. Henry and W. T. Yang, Langmuir, 2009, 25, 10209–10217 CrossRef CAS; Y. J. Song, H. Modrow, L. L. Henry, C. K. Saw, E. E. Doomes, V. Palshin, J. Hormes and C. Kumar, Chem. Mater., 2006, 18, 2817–2827 CrossRef.
- B. F. Cottam, S. Krishnadasan, A. J. deMello, J. C. deMello and M. S. P. Shaffer, Lab Chip, 2007, 7, 167–169 RSC; T. H. Eun, S. H. Kim, W. J. Jeong, S. J. Jeon, S. H. Kim and S. M. Yang, Chem. Mater., 2009, 21, 201–203 CrossRef CAS; M. Takagi, T. Maki, M. Miyahara and K. Mae, Chem. Eng. J., 2004, 101, 269–276 CrossRef; H. Z. Wang, H. Nakamura, M. Uehara, M. Miyazaki and H. Maeda, Chem. Commun., 2002, 1462–1463 RSC.
- S. A. Khan, A. Gunther, M. A. Schmidt and K. F. Jensen, Langmuir, 2004, 20, 8604–8611 CrossRef CAS.
- M. Kawase, T. Suzuki and K. Miura, Chem. Eng. Sci., 2007, 62, 4875–4879 CrossRef CAS.
- K. I. Sotowa, K. Irie, T. Fukumori, K. Kusakabe and S. Sugiyama, Chem. Eng. Technol., 2007, 30, 383–388 CrossRef CAS.
- A. Abou Hassan, O. Sandre, V. Cabuil and P. Tabeling, Chem. Commun., 2008, 1783–1785 RSC; W. B. Lee, C. H. Weng, F. Y. Cheng, C. S. Yeh, H. Y. Lei and G. B. Lee, Biomed. Microdevices, 2009, 11, 161–171 CrossRef CAS; T. Miyake, T. Ueda, N. Ikenaga, H. Oda and M. Sano, J. Mater. Sci., 2005, 40, 5011–5013 CrossRef.
- L. Frenz, A. El Harrak, M. Pauly, S. Begin-Colin, A. D. Griffiths and J.-C. Baret, Angew. Chem., Int. Ed., 2008, 47, 6817–6820 CrossRef CAS; K. Kumar, A. M. Nightingale, S. H. Krishnadasan, N. Kamaly, M. Wylenzinska-Arridge, K. Zeissler, W. R. Branford, E. Ware, A. J. deMello and J. C. deMello, J. Mater. Chem., 2012, 22, 4704–4708 RSC.
- P. H. Hoang, H. Park and D. P. Kim, J. Am. Chem. Soc., 2011, 133, 14765–14770 CrossRef CAS.
- Y. Pan, J. Yao, L. Zhang and N. Xu, Ind. Eng. Chem. Res., 2009, 48, 8471–8477 CrossRef CAS.
- D. Jeevarathinam, A. K. Gupta, B. Pitchumani and R. Mohan, Chem. Eng. J., 2011, 173, 607–611 CrossRef CAS.
- R. Kikkeri, P. Laurino, A. Odedra and P. H. Seeberger, Angew. Chem., Int. Ed., 2010, 49, 2054–2057 CrossRef CAS; H. Z. Wang, X. Y. Li, M. Uehara, Y. Yamaguchi, H. Nakamura, M. P. Miyazaki, H. Shimizu and H. Maeda, Chem. Commun., 2004, 48–49 RSC; H. Z. Wang, H. Nakamura, M. Uehara, Y. Yamaguchi, M. Miyazaki and H. Maeda, Adv. Funct. Mater., 2005, 15, 603–608 CrossRef.
- A. Abou-Hassan, R. Bazzi and V. Cabuil, Angew. Chem., Int. Ed., 2009, 48, 7180–7183 CrossRef CAS.
- S. Duraiswamy and S. A. Khan, Nano Lett., 2010, 10, 3757–3763 CrossRef CAS.
- A. Knauer, A. Thete, S. Li, H. Romanus, A. Csáki, W. Fritzsche and J. M. Köhler, Chem. Eng. J., 2011, 166, 1164–1169 CrossRef CAS.
- C. G. Lee, M. Uehara, H. Nakamura and H. Maeda, J. Chem. Eng. Jpn., 2008, 41, 644–648 CrossRef CAS.
- R. D. Chambers, M. A. Fox, D. Holling, T. Nakano, T. Okazoe and G. Sandford, Lab Chip, 2005, 5, 191–198 RSC.
- N. de Mas, A. Gunther, M. A. Schmidt and K. F. Jensen, Ind. Eng. Chem. Res., 2009, 48, 1428–1434 CrossRef CAS; K. Jahnisch, M. Baerns, V. Hessel, W. Ehrfeld, V. Haverkamp, H. Lowe, C. Wille and A. Guber, J. Fluorine Chem., 2000, 105, 117–128 CrossRef; P. Styring and A. I. R. Parracho, Beilstein J. Org. Chem., 2009, 5, 29 CrossRef.
- Y. Wada, M. A. Schmidt and K. F. Jensen, Ind. Eng. Chem. Res., 2006, 45, 8036–8042 CrossRef CAS.
- T. Iwasaki, N. Kawano and J.-i. Yoshida, Org. Process Res. Dev., 2006, 10, 1126–1131 CrossRef CAS; R. Schenk, V. Hessel, C. Hofmann, J. Kiss, H. Lowe and A. Ziogas, Chem. Eng. J., 2004, 101, 421–429 CrossRef.
- S. L. Poe, M. A. Cummings, M. R. Haaf and D. T. McQuade, Angew. Chem., Int. Ed., 2006, 45, 1544–1548 CrossRef CAS.
- A. M. Nightingale and J. C. de Mello, Adv. Mater., DOI:10.1002/adma.201203252, in press.
- I. Shestopalov, J. D. Tice and R. F. Ismagilov, Lab Chip, 2004, 4, 316–321 RSC.
- N. Gaponik, S. G. Hickey, D. Dorfs, A. L. Rogach and A. Eychmueller, Small, 2010, 6, 1364–1378 CrossRef CAS.
- E. M. Chan, A. P. Alivisatos and R. A. Mathies, J. Am. Chem. Soc., 2005, 127, 13854–13861 CrossRef CAS.
- A. M. Nightingale, S. H. Krishnadasan, D. Berhanu, X. Niu, C. Drury, R. McIntyre, E. Valsami-Jones and J. C. deMello, Lab Chip, 2011, 11, 1221–1227 RSC.
- L. Philpott, The Ultimate Book of Decorative Knots, Skyhorse Publishing Company, 2010 Search PubMed.
- Y. Kikutani, A. Hibara, K. Uchiyama, H. Hisamoto, M. Tokeshi and T. Kitamori, Lab Chip, 2002, 2, 193–196 RSC; J. Yue, R. Boichot, L. Luo, Y. Gonthier, G. Chen and Q. Yuan, AIChE J., 2010, 56, 298–317 Search PubMed.
- M. Al-Rawashdeh, L. J. M. Fluitsma, T. A. Nijhuis, E. V. Rebrov, V. Hessel and J. C. Schouten, Chem. Eng. J., 2012, 181, 549–556 CrossRef CAS; N. de Mas, A. Gunther, T. Kraus, M. A. Schmidt and K. F. Jensen, Ind. Eng. Chem. Res., 2005, 44, 8997–9013 CrossRef; M. Mendorf, H. Nachtrodt, A. Mescher, A. Ghaini and D. W. Agar, Ind. Eng. Chem. Res., 2010, 49, 10908–10916 CrossRef.
- R. E. Bailey and S. M. Nie, J. Am. Chem. Soc., 2003, 125, 7100–7106 CrossRef CAS; L. Liao, H. Zhang and X. Zhong, J. Lumin., 2011, 131, 322–327 CrossRef.
- R. E. Bailey, J. B. Strausburg and S. M. Nie, J. Nanosci. Nanotechnol., 2004, 4, 569–574 CrossRef CAS.
- W. W. Yu, Y. A. Wang and X. G. Peng, Chem. Mater., 2003, 15, 4300–4308 CrossRef CAS.
- A. H. Reshak, I. V. Kityk, R. Khenata and S. Auluck, J. Alloys Compd., 2011, 509, 6737–6750 CrossRef CAS.
- W. Jiang, A. Singhal, J. Zheng, C. Wang and W. C. W. Chan, Chem. Mater., 2006, 18, 4845–4854 CrossRef CAS; B. Xing, W. Li, X. Wang, H. Dou, L. Wang, K. Sun, X. He, J. Han, H. Xiao, J. Miao and Y. Li, J. Mater. Chem., 2010, 20, 5664–5674 RSC.
- J. J. Li, Y. A. Wang, W. Z. Guo, J. C. Keay, T. D. Mishima, M. B. Johnson and X. G. Peng, J. Am. Chem. Soc., 2003, 125, 12567–12575 CrossRef CAS.
- T. Kim, S. W. Kim, M. Kang and S.-W. Kim, J. Phys. Chem. Lett., 2012, 3, 214–218 CrossRef CAS.
- S. H. Choi, E. G. Kim, J. Park, K. An, N. Lee, S. C. Kim and T. Hyeon, J. Phys. Chem. B, 2005, 109, 14792–14794 CrossRef CAS.
- Y. Lee, J. Lee, C. J. Bae, J. G. Park, H. J. Noh, J. H. Park and T. Hyeon, Adv. Funct. Mater., 2005, 15, 503–509 CrossRef CAS.
- J. H. Liu, J. B. Fan, Z. Gu, J. Cui, X. B. Xu, Z. W. Liang, S. L. Luo and M. Q. Zhu, Langmuir, 2008, 24, 5241–5244 CrossRef CAS.
- F.-J. Fan, Y.-X. Wang, X.-J. Liu, L. Wu and S.-H. Yu, Adv. Mater., 2012, 24, 6158–6163 CrossRef CAS.
- T. Y. Yu, J. Joo, Y. I. Park and T. Hyeon, Angew. Chem., Int. Ed., 2005, 44, 7411–7414 CrossRef CAS.
- G. Garnweitner, L. M. Goldenberg, O. V. Sakhno, M. Antonietti, M. Niederberger and J. Stumpe, Small, 2007, 3, 1626–1632 CrossRef CAS.
- J. Park, K. J. An, Y. S. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ta10458c |
| ‡ Note, using separate feed-systems for each channel would entail unacceptable cost and complexity. |
| § Note, there are several reports relating to the successful use of gas/liquid segmented flow reactors for high temperature nanocrystal synthesis. However in these reactors the reagent phase is able to make contact with the channel walls, risking particle deposition and eventual failure of the reactor. |
| ¶ The small volume of the syringes facilitated pulseless delivery of the precursor solution into the reactor against a high back-pressure. |
| || The switchover phase (during which flow through the reactor was momentarily interrupted) lasted approximately 1 s, implying an active duty cycle of >99% for a typical flow rate of 2 ml min−1. |
| ** An alternative option would have been to insert back-pressure valves at the end of each line and adjust their thresholds accordingly. |
| This journal is © The Royal Society of Chemistry 2013 |