 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot, large-scale synthesis of magnetic activated carbon nanotubes and their applications for arsenic removal†
Jie
Ma
a,
Zhiliang
Zhu
a,
Bo
Chen
a,
Mingxuan
Yang
a,
Huiming
Zhou
a,
Chen
Li
a,
Fei
Yu
*ab and
Junhong
Chen
*ac
aState Key Laboratory of Pollution Control and Resource Reuse, School of Environmental Science and Engineering, Tongji University, 1239 Siping Road, Shanghai 200092, P. R. China. E-mail: jma@tongji.edu.cn; Tel: +86-21-6598 1831
bSchool of Environmental Science and Engineering, Shanghai Jiao Tong University, 800 Dong Chuan Road, Shanghai 200240, P. R. China. E-mail: fyu@vip.163.com
cDepartment of Mechanical Engineering, Laboratory for Surface Studies, University of Wisconsin–Milwaukee, Milwaukee, WI 53211, USA. E-mail: jhchen@uwm.edu
First published on 26th February 2013
Abstract
We report a one-pot method to synthesize magnetic iron oxide/CNT composites (MI/CNTs) based on as-prepared CNTs (APCNTs) using KOH activation. MI/CNTs have high specific surface area, good dispersion and magnetic properties, making them promising for use as adsorbents for arsenic removal. The results of this work are highly significant for large-scale applications of APCNTs containing Fe catalytic particles without the need for prior purification.
Arsenic is considered as one of the most hazardous elements for the living organisms, and its presence in natural waters can result in serious environmental problems. The US Environmental Protection Agency (EPA) has set the arsenic standard for drinking water at 10 parts per billion to protect consumers served by public water systems from the effects of long-term chronic exposure to arsenic. This value is related to the total concentration of arsenic, but it was recognized that arsenic toxicity for humans depends strongly on its chemical form. It occurs with valence states of −3, 0, +3 (arsenite, As[III]) and +5 (arsenate, As[V]). The valence states −3 and 0 occur rarely. Arsenic is primarily present in water as arsenite (AsO3)3− and arsenate (AsO4)3−. Despite the availability of many technologies for treating arsenic, their ability to remove arsenic is limited.
Adsorption has been found to be an efficient and economic process for removing arsenic from water, depending on the media used. Several iron oxides, such as granular ferric hydroxide, amorphous hydrous ferric oxide, crystalline hydrous ferric oxide,1 goethite and magnetite,2 and iron/aluminium-based granular materials,3 are well-known adsorbents that efficiently remove arsenic; however, these adsorbents are difficult to use in continuous flow systems due to their small particle size and instability. To overcome this difficulty, several researchers have combined magnetic iron oxides with carbon nanotubes (CNTs). CNTs provide enough adsorption sites for harmful metallic ions, as well as good support for other adsorbent materials.4,5 The present methods for synthesizing iron oxide/CNTs, however, have some critical disadvantages:6 (a) as-prepared CNTs (APCNTs) are usually purified first using acid to remove the metal particles and carbonaceous by-products;7 (b) the purified CNTs need to be modified by functional groups using oxidation methods,8 and then iron oxide nanoparticles will load successfully on the CNTs by various chemical or physical techniques.
Therefore, the existing synthesis methods are expensive, complex, time-consuming, and environmentally unfriendly, leading to a low yield and limited practical applications. Some studies indicated that amorphous iron oxide adsorbents based on akaganeite and amorphous iron hydroxide have higher arsenic adsorption capacities than those based on hematite, goethite, and magnetite. However, it is difficult to separate iron oxide/CNTs from aqueous solutions using a magnet due to the amorphous structure with weak magnetic properties. In addition, there are serious concerns about the health and environmental risks of iron oxide/CNTs once they have been released into the environment.9 These issues may ultimately hinder the wide-spread practical applications of the iron oxide/CNT composites.
In this paper, we report a one-pot method to synthesize magnetic iron oxide/CNT composites (MI/CNTs) using APCNTs modified by KOH activation. This facile one-step synthesis method has the following advantages: (a) iron nanoparticles in the APCNTs can be used directly without any purification; (b) iron oxide can be loaded on the CNTs without further chemical modification; and (c) CNTs are activated with high specific surface area (SSA) and pore volume. Here, MI/CNTs were used as adsorbents to remove arsenic pollutants, which exhibited excellent adsorption and magnetic separation properties. The adsorption capacities are higher than what have been shown in previous reports.10–13 After adsorption, the MI/CNT adsorbents could be effectively and immediately separated using a magnet, thereby reducing the potential risks from MI/CNT nanoparticles as an environmental contaminant. Moreover, improved manufacturing and large-scale production have already caused the price of CVD-produced CNTs to fall substantially; therefore, MI/CNTs may be a promising magnetic adsorbent for removing pollutants using APCNTs.
Fig. 1a displays the transmission electron microscopy (TEM) images of APCNTs; the diameter of APCNTs is about 20–30 nm and the length is about ∼1 μm. After the activation treatment, the structure of APCNTs has been clearly modified and the length of MI/CNTs is obviously shortened. Furthermore, part of the hollow tubular structure is destroyed, large quantities of defects are produced, and many flaky apertures are generated on the surface, as shown in Fig. 1b. The elemental composition of MI/CNTs was analyzed by energy-dispersive X-ray spectroscopy (EDS), as shown in Fig. S1.† The X-ray diffraction (XRD) patterns of MI/CNT hybrids indicated that the MI/CNTs were a mixture of two/three phases: γ-Fe2O3/Fe3O4 and CNTs. Well-resolved diffraction peaks reveal the good crystallinity of γ-Fe2O3/Fe3O4 specimens; no peaks corresponding to impurities were detected. Peaks of C with relatively high intensity and symmetry are clearly observed in Fig. 1c. This observation suggests that the graphite structure remained, even after strong activation reaction; therefore, we may conclude that MI/CNT heterostructures were formed using the KOH-activation method.
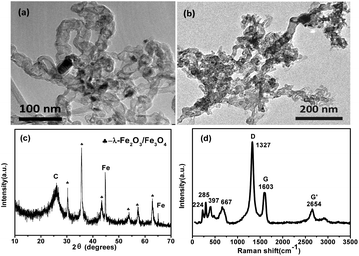 | ||
| Fig. 1 TEM images (a and b) of APCNTs and MI/CNTs and XRD patterns (c) and Raman spectrum (d) of MI/CNTs. | ||
The Raman spectrum of MI/CNTs is shown in Fig. 1d. For MI/CNTs, the remaining peaks at 224 and 285 cm−1 are assigned to the A1g and Eg modes of α-Fe2O3; the peak at 397 cm−1 is assigned to the T2g modes of λ-Fe2O3; and the peak at 667 cm−1 is assigned to the A1g modes of Fe3O4.14 The results indicate that magnetic iron oxide in MI/CNTs may be a mixture phase composed of α-Fe2O3, λ-Fe2O3, and Fe3O4. The G peak at 1603 cm−1 is related to E2g graphite mode.15–17 The D-line at ∼1327 cm−1 is induced by defective structures. The intensity ratio of the G and D peaks (IG/ID) is an indicator for estimating the structure quality of CNTs, as shown in Fig. 1d, which suggests that the structure of the CNTs was destroyed after the KOH-activation process.
In Fig. 2a, the thermogravimetric (TG) analysis of the MI/CNTs exhibits two main weight-loss regions. MI/CNTs are considerably stable and show a little weight loss close to 5% below 200 °C in the first region, which can be attributed to the evaporation of adsorbed water and the elimination of carboxylic groups and hydroxyl groups on the MI/CNTs. The rapid weight-loss region can be due to the oxidation of CNTs. It is clearly seen that the main thermal event temperature was at ∼500 °C; however, the thermal event temperature is so high that MI/CNTs could readily meet the application needs of adsorbents in water treatment.
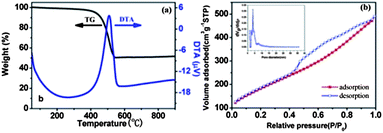 | ||
| Fig. 2 Thermal analysis (a), adsorption/desorption isotherms of N2 and pore distribution (b) of MI/CNTs. | ||
The specific surface area (SSA) and pore-size characterization of MI/CNTs were performed by nitrogen (77.4 K) adsorption/desorption experiments with density functional theory (DFT) methods,18 as shown in Fig. 2b. The MI/CNTs had a high SSA of ∼662.1 m2 g−1 (calculated in the linear relative pressure range from 0.1 to 0.3). The SSA of MI/CNTs is drastically increased by ∼5 times than that of APCNTs; such increases correspond to a decrease in the mean pore diameter from ∼11.03 to ∼2.26 nm (BJH). After alkali-activation treatment, not only the tube tip was opened, but also large quantities of new micropore structures with small sizes were produced. This implies that the MI/CNTs possess more small-pores after the present activation treatment and thus could lead to a higher SSA. The detailed features of the pore-distribution analysis are presented in Table S1.†
Fig. 2b displays the results of the cumulative pore volume and pore size analysis from nitrogen adsorption by applying a hybrid nonlocal DFT (NLDFT) kernel, assuming a slit-shaped pore for the micropores and a cylindrical pore for the mesopores. The obtained pore-size/volume distribution indicates that this MI/CNTs sample is distinctive from APCNTs. Compared with APCNTs, the total PV of MI/CNTs decreased due to the disappearance of macropores. The meso-PV and micro-PV of MI/CNTs improved by almost ∼1.48 and ∼5.73 times than that of APCNTs. The stronger peak of pore distribution of MI/CNTs exists at ∼2 nm, which indicates the presence of plenty of micro-pores after the alkali-activation treatment.
The composition of MI/CNTs was further determined by X-ray photoelectron spectroscopy (XPS), as shown in Fig. 3 and Table S2.† Typical XPS survey scans of the MI/CNTs are shown in Fig. 3a. Fig. 3b shows the principal deconvoluted component of the C1s region recorded for the MI/CNTs. We can see that the strongest peak at 284.6 eV is assigned to double-bonding carbons for CNTs and results from non-functionalized carbon. The peak at the binding energy of about 285.1 eV is a consequence of single-bonding carbon for CNTs.19 The O1s spectra consist of three peaks that are assigned to Fe–O (529.8 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (531.2 eV), and C–O (533.0 eV) bonds,20 which suggests the introduction of new functional groups and the iron oxide nanoparticle loading on the surfaces of MI/CNTs. The Fe2p spectrum (Fig. 3c) shows two broad peaks with satellite peaks at 711.4 eV and 724.5 eV representing Fe2p3/2 and Fe2p1/2, respectively. The presence of these chemical bonds demonstrates that iron oxide nanoparticles formed on the surface of MI/CNTs.
O (531.2 eV), and C–O (533.0 eV) bonds,20 which suggests the introduction of new functional groups and the iron oxide nanoparticle loading on the surfaces of MI/CNTs. The Fe2p spectrum (Fig. 3c) shows two broad peaks with satellite peaks at 711.4 eV and 724.5 eV representing Fe2p3/2 and Fe2p1/2, respectively. The presence of these chemical bonds demonstrates that iron oxide nanoparticles formed on the surface of MI/CNTs.
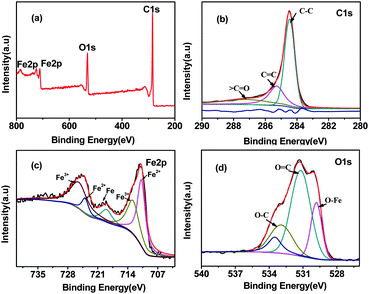 | ||
| Fig. 3 XPS survey scans of the MI/CNTs (a), the C1s deconvolution of MI/CNTs (b) for the Fe2p region of MI/CNTs (c), and the O1s deconvolution of MI/CNTs (d). | ||
The magnetization properties of MI/CNTs were investigated at room temperature by measuring magnetization curves (Fig. 4). The saturation magnetization Ms of MI/CNTs is 27.2 emu g−1 for MI/CNTs (magnetic field = ±10 kOe), indicating that MI/CNTs have high magnetism. The loop of MI/CNTs exhibits very low coercive field (48 Oe) and remanence values (0.87 emu g−1), indicating that MI/CNT hybrids are very close to behaving as superparamagnets at room temperature, which can be beneficial to the reuse without reunite for magnetization. After magnetic separation, the concentration of residual CNTs in an aqueous solution was estimated using a UV-visible absorption-based approach.21,22 It is noteworthy to observe that the concentration of residual CNTs is almost 0 g L−1, as shown in Fig. S2 and S3.†
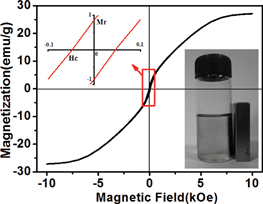 | ||
| Fig. 4 Hysteresis loop of MI/CNTs and the digital photograph of MI/CNTs with magnetic separation. | ||
The MI/CNT powders can be well dispersed in water during adsorption, so the removal of arsenic by MI/CNTs was found to be rapid at the initial period (in the first 1 min) and then became slower (1–10 min). The rate of removal reached a plateau after approximately ∼10 minutes of the experiment, as shown in Fig. S4.† The MI/CNTs' adsorption isotherms for removing As(V) and As(III) are shown in Fig. 5, which indicates that APCNTs have a smaller adsorption capacity of As(V)/As(III), due to poor interaction between CNTs and arsenic pollutants. After the KOH-activation treatment, a larger number of iron oxide nanoparticles were decorated on the surface of MI/CNTs, and the resulting adsorption capacity increased significantly, which suggests that the iron oxides contributed to the increase of adsorption capacity.
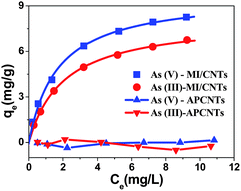 | ||
| Fig. 5 Equilibrium adsorption isotherms of As(V) and As(III) on APCNTs and MI/CNTs. | ||
The equilibrium adsorption of arsenic on MI/CNTs was analyzed using the Langmuir23 and Freundlich24 isotherm models. Fig. S5 shows the isotherms based on the experimental data, and the parameters obtained from linear regression using adsorption models are shown in Table S3.† Based on the determination coefficient (R2), the Langmuir model fits the experimental data better than the Freundlich model. The applicability of the Langmuir isotherm suggests that specific homogenous sites within the adsorbent are involved.25 The computed maximum monolayer capacities have wonderful values of ∼9.74 mg g−1 for As(V) and ∼8.13 mg g−1 for As(III) on the MI/CNTs, which are also higher than those of previously reported adsorbents.10–13 These results suggest that MI/CNTs have great potential for As(V) and As(III) removal. The Dubinin–Radushkevich (D–R)26 isotherm model was applied to distinguish between the physical and chemical adsorption of As(V) and As(III) on MI/CNTs, as shown in Fig. S5c.† The values of E (mean energy of adsorption) exceed 8 kJ mol−1 for As(V) and As(III), suggesting the removal process may follow chemisorption between As(V)/As(III) and MI/CNTs.27 Surface structure information of MI/CNTs was analyzed by XPS after arsenic adsorption. The XPS spectra of As3d are shown in Fig. S6 and Table S2.† The quantitative analysis of As(III) adsorbed MI/CNTs shows 50.8% of As(III) and 49.2% of As(V) on the sorbent surface; however, only As(V) exists on the As(V) adsorbed MI/CNTs. This result indicates solid-state oxidation between arsenate and arsenite on the surface of MI/CNTs. It has been reported that the arsenic adsorption mechanism involves electrostatic attraction28 and surface complexation29,30 between the arsenic species and iron oxides in solution. Arsenate surface complexes possibly exist in three forms of binuclear, bidentate, and monodentate complexes through the ligand exchange reaction.31 After arsenic adsorption, the atomic ratio of Fe on the surface decreased from 6.58% to 5.35% for As(III) and 5.86% for As(V) with the increase of As atomic ratio, indicating that Fe atoms were overlaid by the adsorbed arsenic. The higher adsorption capacity of MI/CNTs can be attributed to the following reasons. First, a large number of iron oxide nanoparticles formed on the surface of MI/CNTs and iron oxide adsorbents have demonstrated superior adsorption performance.32–34 Second, oxygen-containing functional groups of MI/CNTs would improve hydrophilicity and dispersibility in aqueous solutions. For As(V)/As(III) with considerable solubility, a better dispersion of MI/CNTs in water will increase the available adsorption sites, which may be favorable for the aqueous phase adsorption.35 Third, the increased SSA and meso-PV/micro-PV would provide more adsorption sites for As(V)/As(III).36 Therefore, the adsorption properties obviously increased after the KOH activation treatment.
In summary, magnetic MI/CNT hybrids were synthesized by a facile KOH-activation method based on APCNTs. MI/CNTs have high SSA, good dispersibility and desirable magnetic properties, making them promising for use as an adsorbent for As(V) and As(III) removal with excellent adsorption capacity and magnetic separation properties. The results of this work are highly significant for practical large-scale applications of as-prepared single-walled or multi-walled carbon nanotubes containing Fe catalytic particles without the need for prior purification.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21207100, 51072135), Innovation Program of Shanghai Municipal Education Commission (2010L10247150), Fundamental Research Funds for the Central Universities, and sponsored by Shanghai Pujiang Program (10PJ1410200).Notes and references
- W. Xu, H. J. Wang, K. Wu, R. P. Liu, W. X. Gong and J. H. Qu, Water Environ. Res., 2012, 84, 521–528 CrossRef CAS.
- Y. Mamindy-Pajany, C. Hurel, N. Marmier and M. Romeo, Desalination, 2011, 281, 93–99 CrossRef CAS.
- D. E. Giles, M. Mohapatra, T. B. Issa, S. Anand and P. Singh, J. Environ. Manage., 2011, 92, 3011–3022 CrossRef CAS.
- M. Yin, M. L. Wang, F. Miao, Y. X. Ji, Z. Tian, H. B. Shen and N. Q. Jia, Carbon, 2012, 50, 2162–2170 CrossRef CAS.
- Y. J. Fu, L. Y. Zhang and G. Chen, Carbon, 2012, 50, 2563–2570 CrossRef CAS.
- J. Ma, F. Yu, Z. H. Wen, M. X. Yang, H. M. Zhou, C. Li, L. Jin, L. Zhou, L. Chen, Z. W. Yuan and J. H. Chen, Dalton Trans., 2012, 42, 1356–1359 RSC.
- J. Ma and J. N. Wang, Chem. Mater., 2008, 20, 2895–2902 CrossRef CAS.
- F. Yu, J. H. Chen, L. Chen, J. Huai, W. Y. Gong, Z. W. Yuan, J. H. Wang and J. Ma, J. Colloid Interface Sci., 2012, 378, 175–183 CrossRef CAS.
- D. L. Plata, P. M. Gschwend and C. M. Reddy, Nanotechnology, 2008, 19, 185706 CrossRef CAS.
- B. S. Tawabini, S. F. Al-Khaldi, M. M. Khaled and M. A. Atieh, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2011, 46, 215–223 CrossRef CAS.
- S. A. Ntim and S. Mitra, Abstr. Pap., Jt. Conf. – Chem. Inst. Can. Am. Chem. Soc., 2010, 240 Search PubMed.
- S. A. Ntim and S. Mitra, J. Colloid Interface Sci., 2012, 375, 154–159 CrossRef.
- S. A. Ntim and S. Mitra, J. Chem. Eng. Data, 2011, 56, 2077–2083 CrossRef.
- A. M. Jubb and H. C. Allen, ACS Appl. Mater. Interfaces, 2010, 2, 2804–2812 CAS.
- Z. H. Yu and L. E. Brus, J. Phys. Chem. B, 2001, 105, 6831–6837 CrossRef CAS.
- A. G. Souza, A. Jorio, G. G. Samsonidze, G. Dresselhaus, M. A. Pimenta, M. S. Dresselhaus, A. K. Swan, M. S. Unlu, B. B. Goldberg and R. Saito, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 035427 CrossRef.
- A. Gruneis, R. Saito, T. Kimura, L. G. Cancado, M. A. Pimenta, A. Jorio, A. G. Souza, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 155405–155411 CrossRef.
- P. I. Ravikovitch, A. Vishnyakov, R. Russo and A. V. Neimark, Langmuir, 2000, 16, 2311–2320 CrossRef CAS.
- C. C. Chen, C. F. Chen, C. M. Chen and F. T. Chuang, Electrochem. Commun., 2007, 9, 159–163 CrossRef CAS.
- P. C. J. Graat and M. A. J. Somers, Appl. Surf. Sci., 1996, 100, 36–40 CrossRef.
- S. Attal, R. Thiruvengadathan and O. Regev, Anal. Chem., 2006, 78, 8098–8104 CrossRef CAS.
- S. H. Jeong, K. K. Kim, S. J. Jeong, K. H. An, S. H. Lee and Y. H. Lee, Synth. Met., 2007, 157, 570–574 CrossRef CAS.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- H. M. F. Freundlich, J. Phys. Chem., 1906, 57, 385–471 CAS.
- F. Yu, J. H. Chen, M. X. Yang, L. Zhou, L. Jin, C. Su, F. L. Li, L. Chen, Z. W. Yuan, L. L. Yu and J. Ma, New J. Chem., 2012, 36, 1940–1943 RSC.
- B. M. Jovanovic, V. L. Vukasinovic-Pesic and L. V. Rajakovic, Water Environ. Res., 2011, 83, 498–506 CrossRef CAS.
- S. S. Tahir and N. Rauf, Chemosphere, 2006, 63, 1842–1848 CrossRef CAS.
- H. M. Guo, D. Stuben and Z. Berner, Appl. Geochem., 2007, 22, 1039–1051 CrossRef CAS.
- R. J. Donahoe, L. Yang and A. Lanzirotti, Geochim. Cosmochim. Acta, 2005, 69, 616 Search PubMed.
- R. L. Vaughan and B. E. Reed, Water Res., 2005, 39, 1005–1014 CrossRef CAS.
- D. M. Sherman and S. R. Randall, Geochim. Cosmochim. Acta, 2003, 67, 4223–4230 CrossRef CAS.
- M. Gallegos-Garcia, K. Ramirez-Muniz and S. X. Song, Miner. Process. Extr. Metall. Rev., 2012, 33, 301–315 CrossRef CAS.
- D. VanDorn, M. T. Ravalli, M. M. Small, B. Hillery and S. Andreescu, J. Chem. Educ., 2011, 88, 1119–1122 CrossRef CAS.
- X. D. Gao and J. Chorover, Environ. Chem., 2012, 9, 148–157 CrossRef CAS.
- F. Yu, J. Ma and Y. Q. Wu, J. Hazard. Mater., 2011, 192, 1370–1379 CrossRef CAS.
- J. Ma, F. Yu, L. Zhou, L. Jin, M. X. Yang, J. S. Luan, Y. H. Tang, H. B. Fan, Z. W. Yuan and J. H. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 5749–5760 CAS.
Footnote |
| † Electronic supplementary information available: See DOI: 10.1039/c3ta10329c |
| This journal is © The Royal Society of Chemistry 2013 |