 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aromatic polyelectrolytes via polyacylation of pre-quaternized monomers for alkaline fuel cells†
Zhenghui
Zhang
a,
Liang
Wu
a,
John
Varcoe
b,
Chuanrun
Li
a,
Ai Lien
Ong
b,
Simon
Poynton
b and
Tongwen
Xu
*a
aCAS Key Laboratory of Soft Matter Chemistry, School of Chemistry and Material Science, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: twxu@ustc.edu.cn; Fax: +86 0551-3601592; Tel: +86 0551-3601587
bDepartment of Chemistry, University of Surrey, Guildford, GU2 7XH, UK. E-mail: j.varcoe@surrey.ac.uk; Fax: +44 1483-686851; Tel: +44 1483-686838
First published on 18th December 2012
Abstract
To overcome alkali-resistant and synthetic hurdles to alkaline anion-exchange membranes (AAEMs) for alkaline fuel cells, the polyacylation of pre-quaternized monomers as a straightforward and versatile approach has been proposed for the first time. Via this approach, novel aromatic anion-exchange polyelectrolytes featuring a long pendent spacer (i.e., –O–(CH2)4–) instead of a conventional benzyl-type spacer (i.e., –CH2–) are successfully synthesized, and exhibit not only high OH− and CO32− conductivity (91 mS cm−1 and 51 mS cm−1 at 60 °C, respectively) but also outstanding alkaline stability (e.g., no degradation of ammonium groups after aging in 6 mol dm−3 NaOH at 60 °C for 40 days). Using this kind of AAEM, a promising peak power density of 120 mW cm−2 is obtained on a preliminary H2/O2 single cell at 50 °C. This powerful synthetic approach together with exceptional membrane properties should pave the way to the practical application of this kind of AAEMs in alkaline fuel cells.
Introduction
Alkaline fuel cells (AFCs) based on anion-exchange membranes (AEMs) are attracting increasing international attention,1–3 which is greatly boosted by the great promise of using non-noble metal catalysts.4,5 However, on the way to the practical application of AEM-based AFCs, the long-term alkaline stability of anion-exchange groups in AEMs remains one of the most challenging issues. The elevated operation temperature of AFCs can offer three key benefits:1 (1) enhancing the electro-kinetics at the electrodes, (2) improving ionic conductivity of AEMs to reduce resistance, and (3) reducing thermodynamic voltage loss due to pH difference across the AEMs caused by CO2; unfortunately, however, traditional AEMs bearing benzyl-type quaternary ammonium groups are vulnerable to attack from OH− anions in a caustic environment, especially at elevated temperatures.6,7 Recently, guanidinium,8 phosphonium9,10 and bis(terpyridine) Ru(II) groups11 have been incorporated into AEMs as novel anion-exchange groups, yet their long-term alkaline stability at high temperature remains unproven, regardless of their high cost. To date, few AEMs have been reported to withstand such accelerated test conditions as ≥1 mol dm−3 NaOH or KOH (aq.), ≥60 °C and ≥1000 h. Historic work on anion-exchange resins (AERs) by Tomoi et al.12 demonstrated that crosslinked polystyrene-based AERs with long alkylene or alkyleneoxymethylene spacers (at least four carbon atoms) between the benzene ring and the trimethylammonium head-group exhibited much higher alkaline stability than commercial ones with the –CH2– spacer, indicating that long spacers (at least four carbon atoms) could greatly enhance the alkaline stability of alkyltrimethyl ammonium groups. Surprisingly, this highly promising work in the field of AERs has attracted little attention in the field of AEMs.On the other hand, from the viewpoint of structure–morphology–property relationships in the field of proton-exchange membranes (PEMs),13–15 aromatic proton-exchange polyelectrolytes (APEPs), especially side-chain-type APEPs, are among the most promising candidates due to their comb-like structures similar to that of Nafion®. Consequently, analogous to side-chain-type APEPs, side-chain-type aromatic anion-exchange polyelectrolytes (AAEPs), i.e., aromatic polymers bearing pendent anion-exchange groups instead of cation-exchange groups along the aromatic main-chain via various spacers, are expected to be very promising materials for AEMs. However, most AAEPs are synthesized via multi-step post-modification, e.g. the chloromethylation–quaternization route4–10,16–21 or the bromomethylation–quaternization route.22–24 Both routes can only lead to benzyl-type AEMs, which may hardly be considered as side-chain-type AAEPs since the –CH2– spacer is too short.
Recently, a facile and versatile route to side-chain-type APEPs based on the concept of direct polyacylation of pre-sulfonated monomers has been successfully developed by our group,25 which directs us to extend its scope to the preparation of side-chain-type AAEPs based on the concept of direct polyacylation of pre-quaternized monomers (Scheme S1 in ESI†), where the length of spacer and the species of cationic group can be widely varied. As an example, the preparation of AAEPs with pendent O-butyl trimethyl ammonium groups (OBuTMA–AAEPs) via the direct polyacylation of a pre-quaternized monomer is reported herein. The properties of the corresponding AEMs in terms of alkaline stability, conductivity, water uptake (WU), linear swelling ratio (LSR) as well as single cell performance were fully evaluated.
Experimental
Materials
2,2′-Dihydroxybiphenyl, 4,4′-oxydibenzoic acid (ODBA), and trifluoromethane sulfonic acid (TFSA) were purchased from Energy-chemical Inc. (China). K2CO3, acetonitrile, dimethylformaldehyde (DMF), dimethyl sulfoxide (DMSO) and other solvents were supplied by Shanghai Sinopham Chemical Reagent Co. Ltd (China). All these reagents were used as received unless otherwise stated. 2,2′-Dimethoxybiphenyl (DMBP)26 and (4-bromobutyl)trimethylammonium bromide27 were prepared according to the literature.Synthesis of QBP monomer
To a flask equipped with a condenser and a N2 inlet were added CH3CN (80 mL), 2,2′-dihydroxybiphenyl (1.86 g, 10 mmol), K2CO3 (3.45 g, 25 mmol) and (4-bromobutyl)trimethylammonium bromide (5.50 g, 20 mmol). The reaction mixture was stirred at 75 °C for 18 h and then filtered, and the filtrate was dried by rotary evaporation. The thus-obtained white powder was crystallized from CH3CN to obtain the pre-quaternized monomer 2,2'-bis[4-(N,N′,N′′-trimethylammonium)butyloxy]biphenyl dibromide (named as QBP for short) in 86% yield. 1H NMR (400 MHz, D2O) δ (ppm): 7.41 (Ar–H4, t, 2H), 7.27 (Ar–H6, d, 2H), 7.09–7.15 (Ar–H3 and Ar–H5, m, 4H), 4.03 (OCH2, t, 4H), 3.06 (CH2N, m, 4H), 2.88 (CH3N, s, 18H), 1.63 (CH2CH2CH2 CH2, m, 8H).Synthesis of polymer OBuTMA–AAEPs-x
To a flask equipped with a CaCl2 tube were added DMBP (0.2143 g, 1.0 mmol), QBP (0.5744 g, 1.0 mmol), ODBA (0.5165 g, 2.0 mmol) and trifluoromethane sulfonic acid (5.00 mL). The reaction mixture was stirred at 60 °C for 16 h. The viscous reddish solution was dropped slowly into cold water to obtain a fibrous precipitate. The thus-obtained polymer was washed thoroughly with water, followed by drying in vacuo to obtain OBuTMA–AAEPs-1.0 of CF3SO3− type in essentially quantitative yield. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.92–7.13 (Ar–H), 4.16 (OCH2), 3.85 (OCH3), 3.24 (CH2N), 2.97 (CH3N), 1.69 (CH2CH2CH2 CH2). OBuTMA–AAEPs-1.2 was prepared in the same manner but at a molar ratio (x) of 1.2 between QBP and DMBP.Preparation of membranes
A solution of OBuTMA–AAEPs-x (CF3SO3− type, 8 wt% in DMF) was cast onto a clean and flat glass plate, followed by heating at 60 °C for 24 h. The resulting membrane was then peeled off by immersing in de-ionized water. The thickness of the membranes was in the range of 40–60 μm. To obtain Cl−, OH− and CO32− type membranes, the membrane was anion-exchanged in 1 mol dm−3 aqueous NaCl, NaOH and Na2CO3, respectively.Measurements
1H NMR spectra were obtained on a Bruker AVANCE 400 using D2O or DMSO-d6 (with tetramethylsilane as an internal reference) as a solvent. FT-IR spectra were recorded on a Bruker Vector 22. The thermal behavior was analyzed with a Shimadzu TGA-50H analyzer at a heating rate of 10 °C min−1 under a nitrogen atmosphere. The inherent viscosity was determined using a typical Ubbelohde method with a polymer concentration of 0.5 g dL−1 in DMF at 25 °C. The mechanical properties of the membrane in the hydrated state were measured using an Instron universal tester (Model 3340) at 25 °C at a crosshead speed of 10 mm min−1. Tensile strength (TS) and elongation at break (Eb) values were recorded.Tapping mode atomic force microscopy (AFM) observations were performed with a Veeco diInnova SPM, using micro-fabricated cantilevers with a force constant of approximately 20 N m−1. All Cl− type samples were measured under ambient conditions (25 °C, 50% RH).
Water uptake (WU) and linear swelling ratios (LSR) were calculated by measuring the change of weight and length of the membranes before and after being immersed in water at a given temperature for 24 h according to eqn (1) and (2), respectively.
 | (1) |
 | (2) |
The ionic conductivity of the membranes was measured using a standard four-electrode AC impedance technique on a PGSTAT302N autolab (Metrohm China Ltd). The impedance was determined using a galvanostatic mode with an AC current amplitude of 0.1 mA over a frequency range of 106 Hz to 50 Hz. Using a Bode type plot, the frequency region over which the impedance had a constant value was checked, and the corresponding resistance (R) was obtained from a Nyquist plot. The in-plane conductivity (σ) was calculated according to the following equation:
 | (3) |
Accelerated long-term alkaline stability test: a small piece of membrane (Cl− type, about 20 mg) was immersed in a 30 mL vial full of aqueous NaOH solution (1 mol dm−3 or 6 mol dm−3). The vial was tightly sealed in order to minimize the effect of air and stored at a constant temperature for the desired period. Then the membrane was washed with water, dilute HCl and water successively. After drying, the membrane (Cl− type) was subjected to 1H NMR characterization.
Membrane electrode assembly (MEA) preparation and fuel cell performance: the MEA was fabricated as previously reported except that CO32− type OBuTMA–AAEPs-1.0 was used as the membrane.28 Firstly, a catalyst ink was prepared by mixing HISPEC 3000 catalyst (20% mass Pt/Vulcan XC-72R, Johnson Matthey, UK), poly(vinylbenzyl chloride) (15% mass with respect to the mass of Pt/C) and ethyl acetate. For both the anode and cathode, the ink was carefully sprayed onto wet-proofed carbon paper until a Pt loading of 0.40 mg cm−2 was reached. The two ink-treated electrodes were subsequently immersed overnight in undiluted N,N,N′,N′-tetramethylhexane-1,6-diamine (99%, Acros Organics), then soaked in aqueous K2CO3 (1 mol dm−3) for 2 h (the solution was refreshed twice to ensure complete ion-exchange) and then rinsed intensively with Millipore-grade water (pre-purged with N2 gas). The CO32− type OBuTMA–AAEPs-1.0 AEM was then sandwiched between the two electrodes with the catalyst layer facing the membrane (no hot-pressing was used). The performance of the MEA with an active area of 5.3 cm2 was measured using an 850e fuel cell test station (Scribner Associates, USA). The input flow rates of the anode H2 and cathode O2 were both controlled at 0.6 dm3 min−1. The humidification temperatures of the anode and cathode were controlled to achieve the desired relative humidity. Prior to recording the polarization curve, the MEA was activated by operating the test cell at open circuit voltage (OCV) for 1 h, followed by a potentiostatic discharge at 0.1 V and 0.5 V for 30 min, respectively.
Results and discussion
Synthesis and characterization of the quaternized monomer and polymers
To verify the concept of ‘direct polyacylation of pre-quaternized monomers’ as illustrated in Scheme S1,† QBP, as a representative pre-quaternized diarene monomer, was firstly synthesized via a one-step etherification reaction in 86% yield (Scheme 1a). Then, QBP was copolymerized with 2,2′-dimethoxybiphenyl (DMBP) and 4,4′-oxydibenzoic acid (ODBA) in trifluoromethane sulfonic acid (TFSA) medium to obtain OBuTMA–AAEPs-x, where x (x = 1.0, 1.2) was the molar ratio between QBP and DMBP (Scheme 1b). To the best of our knowledge, this is the first example of the synthesis of aromatic AEMs by directly using monomers with pendent quaternary ammonium groups, although aliphatic AEMs synthesized by directly using pre-quaternized vinyl monomers have been reported.29–32 The polyacylation using TFSA as both the catalyst and reaction medium went smoothly at 60 °C and the homogeneous reaction solution became very viscous within 24 h, indicating the production of polymers with high molecular weight. The thus-obtained OBuTMA–AAEPs-x were of CF3SO3− type because of the evolution of HBr gas at the beginning of the polymerization. The whole procedure, including the synthesis of the QBP monomer, was very simple, facile and scalable. | ||
| Scheme 1 Schematic synthesis of (a) the QBP monomer and (b) the OBuTMA–AAEPs-x polymer (x = 1.0, 1.2). | ||
The structures of OBuTMA–AAEPs-x were confirmed by FT-IR and 1H NMR. Within the region 1640–1730 cm−1 in the FT-IR spectra (Fig. 1), there was a strong characteristic peak (1649 cm−1) assigned to a carbonyl group, but no detectable absorption assigned to a carboxylic acid group, indicating the completion of the polyacylation.
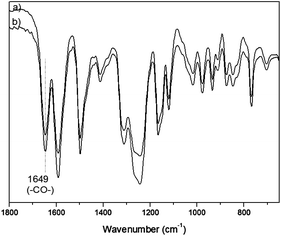 | ||
| Fig. 1 FT-IR spectra of CF3SO3− type OBuTMA–AAEPs-x, (a) x = 1.0; (b) x = 1.2. | ||
1H NMR spectra further verified the structures of OBuTMA–AAEPs-x quantitatively. As seen from Fig. 2, within the region 4.3–1.4 ppm, all peaks were well-assigned, allowing for the accurate calculation of x as well as the ion-exchange capacity (IEC) of OBuTMA–AAEPs-x, based on the integration ratio between peak f (3.85 ppm, OCH3) and peak a (4.16 ppm, OCH2) according to the following equations:
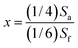 | (4) |
 | (5) |
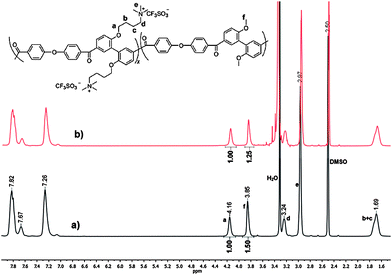 | ||
| Fig. 2 1H NMR spectra of CF3SO3− type OBuTMA–AAEPs-x, (a) x = 1.0; (b) x = 1.2. | ||
OBuTMA–AAEPs-x (CF3SO3− type) also exhibited good solubility in aprotic dipolar solvents such as DMF and DMSO. The DMF solutions of OBuTMA–AAEPs-x (CF3SO3− type) showed high inherent viscosity (ηinh) of more than 0.6 dL g−1 (Table 1), implying their high molecular weights.
Mechanical and thermal properties
The OBuTMA–AAEPs-x membranes also exhibited good mechanical strength. After immersion in water at 60 °C for 48 h, OBuTMA–AAEPs-x (OH−) membranes still exhibited tensile strength (TS) higher than 20 MPa, and elongation at break (Eb) of more than 100% in the hydrated state (Table 1), showing that these membranes were both tough and ductile.The thermal gravimetric (TG) curve of OBuTMA–AAEPs-1.0 (CF3SO3− type) is depicted in Fig. 3. Remarkably, CF3SO3− type OBuTMA–AAEPs-1.0 exhibited high thermal stability up to 338 °C, where the ammonium groups started to decompose. Such a high thermal stability of the ammonium groups should be attributed to the CF3SO3− counter-ion, and is promising for the production of OBuTMA–AAEPs-x (CF3SO3− type) membranes via melt-extrusion, the method adopted in the industrial production of the commercial Nafion® membrane.
 | ||
| Fig. 3 TG curve of CF3SO3− type OBuTMA–AAEPs-1.0. | ||
Long-term alkaline stability
Because of the instability of anion-exchange groups in the state-of-the-art AEMs for AFCs, the long-term alkaline stability of anion-exchange groups should be the first consideration for the development of advanced AEMs for AFCs. Usually, the alkaline stability of AEMs is evaluated by the change in conductivity and IEC by titration after accelerated tests in alkaline solution. However, the change in conductivity may not actually reflect the change of the chemical structures of AEMs, considering that physical changes during accelerated tests could also affect conductivity,33 while titration is a technique of relatively high uncertainty.34 Thanks to the high resolution of the chemical shifts related to the pendent ammonium groups in 1H NMR spectra of OBuTMA–AAEPs-x (Fig. 2), 1H NMR spectrometry was considered a more reliable and accurate technique to reveal the changes in the ammonium groups after accelerated tests.As shown in Fig. 4, for the OBuTMA–AAEPs-1.0 membrane, after soaking in 6 mol dm−3 NaOH at 60 °C for 40 days, there was no observable change in the 1H NMR spectrum of OBuTMA–AAEPs-1.0, except that peak e (3.24 ppm, assigned to CH2N) was overlapped by a water peak due to the exchange of the CF3SO3− counterion with Cl−, that is, almost 100% of the O-butyl trimethyl ammonium (OBuTMA) groups were maintained. In 1 mol dm−3 NaOH at 85 °C for 40 days, 87.4% of the ammonium groups could survive (calculated from the change of the integration ratio of peak e to peak a). Nevertheless, the OBuTMA groups of OBuTMA–AAEPs-1.0 still exhibited higher alkaline stability than most anion-exchange groups reported previously. For example, only about 87% of the benzyl-type trimethylammonium (BzTMA) groups of block poly(arene ether) QPE-X16Y11 (OH− type) remained (from 1.65 to 1.44 mmol g−1 calculated by 1H NMR) after soaking in pure water at 80 °C for 500 h,5 which indicated that the OBuTMA group was much more stable than the BzTMA group, considering that the quaternary ammonium groups would degrade much more quickly at a higher concentration of OH− and higher temperature.7 In comparison with other kinds of anion-exchange groups, Zhang et al.8 reported that poly(aryl ether sulfone) with benzyl-type guanidinium groups maintained its conductivity after immersion in 1 mol dm−3 NaOH at 60 °C for 48 h; Kim et al.35 reported that only about 75% of the phenyl guanidinium groups of poly(aryl ether sulfone) remained (from 1.03 to 0.77 mmol g−1 calculated by 1H NMR) after soaking in 0.5 mol dm−3 NaOH at 80 °C for 382 h; Qu et al.21 reported that the conductivity of poly(aryl ether sulfone) with benzyl-type imidazolium groups decreased by 23.3% after immersion in 3 mol dm−3 KOH at 60 °C for only 24 h; Yan et al.10 mentioned that crosslinked poly(aryl ether sulfone) with benzyl-type phosphonium groups maintained its conductivity after immersion in 1 mol dm−3 KOH at 60 °C or 5 mol dm−3 KOH at room temperature for one month, but the accelerated test conditions were less harsh than those adopted here.
 | ||
| Fig. 4 1H NMR spectra of OBuTMA–AAEPs-1.0: (a) pristine CF3SO3− type sample, (b) Cl− type, after immersion in 6 mol dm−3 NaOH at 60 °C for 40 days, and (c) Cl− type, after immersion in 1 mol dm−3 NaOH at 85 °C for 40 days. | ||
The exceptional alkali-resistance of the OBuTMA groups can firstly be attributed to the lack of conjugation or induction substituents to stabilize possible degradation residues e.g., alcohols resulting from nucleophilic displacement or alkenes from a Hofmann elimination (see detailed analysis in ESI†), thus minimizing possible degradation;2,3,6 secondly, the local micro-environment of ammonium groups within the OBuTMA–AAEPs-1.0 membrane could also play a big role, according to Pratt's calculation36 that better solvation of OH− would lead to slower degradation of ammonium molecules, thus stimulating further investigation into the morphology of the OBuTMA–AAEPs-1.0 membrane.
Morphology characterization
By the virtue of hydrophilic/hydrophobic phase contrast in phase images, tapping mode atomic force microscopy (TM-AFM) has proved to be a useful tool to examine the nanophase formed by sulfonate groups within the Nafion® membrane, as well as other proton-exchange membranes.37–39 TM-AFM can be conducted under ambient conditions, avoiding staining the samples with heavy metals, therefore TM-AFM was used to detect the nanophase formed by ammonium groups within the OBuTMA–AAEPs-1.0 membrane.As shown in Fig. 5, the dark phase, assembled into well-connected nanochannels (about 5–10 nm wide), was assigned to the hydrophilic zone which was probably made up of water as well as ammonium headgroups with counterions, while the bright phase was assigned to the hydrophobic matrix which was mainly made up of aromatic main-chains. Such hydrophilic/hydrophobic phase separation should originate from the Nafion-like side-chain type chemical structure of OBuTMA–AAEPs-1.0. Therefore, most of the ammonium groups and counterions (OH−) of OBuTMA–AAEPs-1.0 were thought to be ‘bathed’ in these hydrophilic channels, where OH− can become well-solvated, thus contributing to the high alkaline stability of the ammonium groups within the OBuTMA–AAEPs-1.0 membrane as mentioned above.
 | ||
| Fig. 5 TM-AFM phase image of Cl− type OBuTMA–AAEPs-1.0. Scale: 500 × 500 nm. | ||
WU, LSR and conductivity properties
As a bridge between chemical structures and physical properties, the above-mentioned phase-segregated micro-structure also plays a crucial role in the control of ionic conductivity, water uptake (WU) and linear swelling ratio (LSR) of OBuTMA–AAEPs-x membranes. As seen from Table 2, while maintaining a reasonable WU as well as low swelling (no more than 25% swelling at 60 °C), OBuTMA–AAEPs-1.2 exhibited an OH− conductivity as high as 91 mS cm−1 at 60 °C, which was much higher than that of the benzyl-trimethylammonium-type random aromatic AEM (rQPE, 35 mS cm−1),17 slightly higher than that of the benzyl-guanidinium-type random aromatic AEM (PSGOH-1.2, 74 mS cm−1)8 and even comparable to that of the benzyl-trimethylammonium-type block aromatic AEM (QPE-X8Y8, 90 mS cm−1).5 Since these membranes all have similar IEC, the high conductivity as well as low swelling of OBuTMA–AAEPs-x could be attributed to the channel-like micro-structure shown in Fig. 5, while for the benzyl-type aromatic AEMs, the spacer (–CH2–) might be too short to induce effective hydrophobic/hydrophilic micro-phase separation. Notably, CO32− conductivity, a physical property that has been long-ignored but might be very important in AFCs as implied by our recent research,40 was also comparatively high. For example, the CO32− conductivity of OBuTMA–AAEPs-1.0 was 24 mS cm−1 at 25 °C and 51 mS cm−1 at 60 °C, while that of a crosslinked aliphatic AEM reported by Coates et al.30 was only 28 mS cm−1 at 50 °C, despite a high OH− conductivity of 111 mS cm−1.| Membranes | IEC (mmol g−1) | Conductivity (mS cm−1) | WU (%) | LSR (%) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| 25 °C | 60 °C | 25 °C | 60 °C | 25 °C | 60 °C | ||||
| a Measured at 30 °C. | |||||||||
| OBuTMA–AAEPs-1.0 (OH−) | 1.75 | 37 | 75 | 40 | 75 | 8.5 | 19.1 | This work | |
| OBuTMA–AAEPs-1.0 (CO32−) | — | 24 | 51 | — | — | — | — | This work | |
| OBuTMA–AAEPs-1.2 (OH−) | 1.87 | 30 | 91 | 62 | 95 | 14.7 | 22.7 | This work | |
| OBuTMA–AAEPs-1.2 (CO32−) | — | 23 | 45 | — | — | — | — | This work | |
| rQPE (OH−) | 1.88 | — | 35 | 95a | — | — | 17 | ||
| QPE-X8Y8 (OH−) | 1.83 | — | 90 | 92a | — | — | 5 | ||
| PSGOH-1.2 (OH−) | 1.89 | — | 74 | — | 107 | — | 46 | 8 | |
Single cell performance
A preliminary single cell test using a membrane electrode assembly (MEA) containing the OBuTMA–AAEPs-1.0 membrane was also carried out. As shown in Fig. 6, under the test conditions of 50 °C and 90% relative humidity (RH) using H2/O2 as reactant gases, the open circuit voltage (zero external current flow) was 1.04 V, indicating that crossover of the reactant gases was very low. Meanwhile, a promising peak power density of 120 mW cm−2 was obtained at 0.4 V. As there are so many factors affecting a cell system, we believe there is a lot of room to optimize AFCs using OBuTMA–AAEPs-x AEMs. | ||
| Fig. 6 H2–O2 polarization curve (filled symbols) and power density curve (empty symbols) of a solid-state AFC at 50 °C containing OBuTMA–AAEPs-1.0 (CO32− type) as the membrane. | ||
Conclusions
In summary, OBuTMA–AAEPs-x were successfully synthesized via the one-step polyacylation of the pre-quaternized monomer QBP. The OBuTMA groups appended along the aromatic main-chain proved to be highly stable in alkaline solution. Such a side-chain-type structure induced channel-like hydrophilic/hydrophobic microphase separation which promoted OH− and CO32− ion transport while maintaining good dimensional stability and mechanical strength. In addition, the flexibility of tuning the structures of main-chains and side-chains via the proposed polyacylation approach will allow for further improvement of the properties of this class of AEMs. The optimization and long-term operation of the AFC single cell is ongoing.Acknowledgements
We acknowledge financial support from the National Basic Research Program (no. 2012CB932800), the National Natural Science Foundation of China (no. 51273185, 21025626, 20974106), the China Postdoctoral Science Foundation (no. 2012M521234), and the UK's Engineering and Physical Sciences Research Council (grants EP/I004882/1 and EP/H025340/1).Notes and references
- J. R. Varcoe and R. C. T. Slade, Fuel Cells, 2005, 5, 187–200 CrossRef CAS.
- K. Nijmeijer, G. Merle and M. Wessling, J. Membr. Sci., 2011, 377, 1–35 CrossRef.
- G. Couture, A. Alaaeddine, F. Boschet and B. Ameduri, Prog. Polym. Sci., 2011, 36, 1521–1557 CrossRef CAS.
- S. Lu, J. Pan, A. Huang, L. Zhuang and J. Lu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20611–20614 CrossRef CAS.
- K. Miyatake, M. Tanaka, K. Fukasawa, E. Nishino, S. Yamaguchi, K. Yamada, H. Tanaka, B. Bae and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 10646–10654 CrossRef.
- E. N. Komkova, D. F. Stamatialis, H. Strathmann and M. Wessling, J. Membr. Sci., 2004, 244, 25–34 CrossRef CAS.
- T. Sata, M. Tsujimoto, T. Yamaguchi and K. Matsusaki, J. Membr. Sci., 1996, 112, 161–170 CrossRef CAS.
- J. Wang, S. Li and S. Zhang, Macromolecules, 2010, 43, 3890–3896 CrossRef CAS.
- S. Gu, R. Cai, T. Luo, Z. Chen, M. Sun, Y. Liu, G. He and Y. Yan, Angew. Chem., Int. Ed., 2009, 48, 6499–6502 CrossRef CAS.
- S. Gu, R. Cai, T. Luo, K. Jensen, C. Contreras and Y. Yan, ChemSusChem, 2010, 3, 555–558 CrossRef CAS.
- Y. Zha, M. L. Disabb-Miller, Z. D. Johnson, M. A. Hickner and G. N. Tew, J. Am. Chem. Soc., 2012, 134, 4493–4496 CrossRef CAS.
- M. Tomoi, K. Yamaguchi, R. Ando, Y. Kantake, Y. Aosaki and H. Kubota, J. Appl. Polym. Sci., 1997, 64, 1161–1167 CrossRef CAS.
- J. E. McGrath, M. A. Hickner, H. Ghassemi, Y. S. Kim and B. R. Einsla, Chem. Rev., 2004, 104, 4587–4611 CrossRef.
- M. Ueda, T. Higashihara and K. Matsumoto, Polymer, 2009, 50, 5341–5357 CrossRef.
- T. J. Peckham and S. Holdcroft, Adv. Mater., 2010, 22, 4667–4690 CrossRef CAS.
- S. Gu, R. Cai and Y. Yan, Chem. Commun., 2011, 47, 2856–2858 RSC.
- M. Tanaka, M. Koike, K. Miyatake and M. Watanabe, Macromolecules, 2010, 43, 2657–2659 CrossRef CAS.
- J. Pan, S. Lu, Y. Li, A. Huang, L. Zhuang and J. Lu, Adv. Funct. Mater., 2010, 20, 312–319 CrossRef CAS.
- J. Pan, Y. Li, L. Zhuang and J. Lu, Chem. Commun., 2010, 46, 8597–8599 RSC.
- M. Tanaka, M. Koike, K. Miyatake and M. Watanabe, Polym. Chem., 2011, 2, 99 RSC.
- F. Zhang, H. Zhang and C. Qu, J. Mater. Chem., 2011, 21, 12744–12752 RSC.
- Y. Wu, C. Wu, T. Xu, X. Lin and Y. Fu, J. Membr. Sci., 2009, 338, 51–60 CrossRef CAS.
- M. R. Hibbs, C. H. Fujimoto and C. J. Cornelius, Macromolecules, 2009, 42, 8316–8321 CrossRef CAS.
- J. Yan and M. A. Hickner, Macromolecules, 2010, 43, 2349–2356 CrossRef CAS.
- Z. Zhang, L. Wu and T. Xu, J. Mater. Chem., 2012, 22, 13996–14000 RSC.
- N. Yonezawa, S. Miyata, T. Nakamura, S. Mori, Y. Ueha and R. Katakai, Macromolecules, 1993, 26, 5262–5263 CrossRef CAS.
- R. A. Bartsch, W. Zhao and Z. Y. Zhang, Synth. Commun., 1999, 29, 2393–2398 CrossRef CAS.
- J. R. Varcoe, R. C. Slade and E. Lam How Yee, Chem. Commun., 2006, 1428–1429 RSC.
- B. Qiu, B. Lin, L. Qiu and F. Yan, J. Mater. Chem., 2012, 22, 1040–1045 RSC.
- N. J. Robertson, H. A. Kostalik, T. J. Clark, P. F. Mutolo, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 3400–3404 CrossRef CAS.
- T. J. Clark, N. J. Robertson, H. A. Kostalik IV, E. B. Lobkovsky, P. F. Mutolo, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2009, 131, 12888–12889 CrossRef CAS.
- H. A. Kostalik, T. J. Clark, N. J. Robertson, P. F. Mutolo, J. M. Longo, H. D. Abruña and G. W. Coates, Macromolecules, 2010, 43, 7147–7150 CrossRef CAS.
- Y. S. Kim, L. Dong, M. A. Hickner, B. S. Pivovar and J. E. McGrath, Polymer, 2003, 44, 5729–5736 CrossRef CAS.
- E. Moukheiber, G. De Moor, L. Flandin and C. Bas, J. Membr. Sci., 2012, 389, 294–304 CrossRef CAS.
- D. S. Kim, A. Labouriau, M. D. Guiver and Y. S. Kim, Chem. Mater., 2011, 23, 3795–3797 CrossRef CAS.
- S. Chempath, B. R. Einsla, L. R. Pratt, C. S. Macomber, J. M. Boncella, J. A. Rau and B. S. Pivovar, J. Phys. Chem. C, 2008, 112, 3179–3182 CAS.
- P. J. James, T. J. McMaster, J. M. Newton and M. J. Miles, Polymer, 2000, 41, 4223–4231 CrossRef CAS.
- P. J. James, M. Antognozzi, J. Tamayo, T. J. McMaster, J. M. Newton and M. J. Miles, Langmuir, 2001, 17, 349–360 CrossRef CAS.
- J. Pang, H. Zhang, X. Li and Z. Jiang, Macromolecules, 2007, 40, 9435–9442 CrossRef CAS.
- L. A. Adams, S. D. Poynton, C. Tamain, R. C. T. Slade and J. R. Varcoe, ChemSusChem, 2008, 1, 79–81 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ta01178f |
| This journal is © The Royal Society of Chemistry 2013 |