 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioinspired materials that self-shape through programmed microstructures
André R.
Studart
*a and
Randall M.
Erb
b
aComplex Materials, Department of Materials, ETH Zurich, 8093 Zurich, Switzerland. E-mail: andre.studart@mat.ethz.ch
bDepartment of Mechanical and Industrial Engineering, Northeastern University, Boston, Massachusetts 02115, USA
First published on 30th August 2013
Abstract
Nature displays numerous examples of materials that can autonomously change their shape in response to external stimuli. Remarkably, shape changes in biological systems can be programmed within the material's microstructure to enable self-shaping capabilities even in the absence of cellular control. Here, we revisit recent attempts to replicate in synthetic materials the shape-changing behavior of selected natural materials displaying deliberately tuned fibrous architectures. Simple processing methods like drawing, spinning or casting under magnetic fields are shown to be effective in mimicking the orientation and spatial distribution of reinforcing fibers of natural materials, thus enabling unique shape-changing features in synthetic systems. The bioinspired design and creation of self-shaping microstructures represent a new pathway to program shape changes in synthetic materials. In contrast to shape-memory polymers and metallic alloys, the self-shaping capabilities in these bioinspired materials originate at the microstructural level rather than the molecular scale. This enables the creation of programmable shape changes using building blocks that would otherwise not display the intrinsic molecular/atomic phase transitions required in conventional shape-memory materials.
Introduction
Biological and synthetic materials that change shape upon external stimuli are of great scientific interest and have also been exploited in a range of different applications in biomedicine, aerospace, transportation and materials science.1,2 Here, we consider these materials to be “self-shaping” when at least one shape change results from internal stresses developed within the material's structure rather than from externally imposed mechanical stresses. Synthetic shape-changing systems, known as shape-memory materials, typically exhibit phase transitions at the molecular and/or atomic scale which enable kinetic trapping of a temporary shape that is later restored to the original conformation under a selective thermal, chemical, magnetic or optical trigger.1–7 In shape-memory polymers, for example, glass transition, crystallization and melting are often used as molecular phase transitions responsible for the memory effect. Similarly, the shape-memory behavior of NiTi metallic alloys relies on the martensitic transformation of the crystalline structure of the metal at a well-defined temperature.6In addition to materials with inherent phase transitions, synthetic systems may also exhibit a stimulated shape change if built from multiple components and layers displaying different responses to certain external stimuli. For example, joining two layers of metals exhibiting distinct thermal expansion coefficients leads to thermostats that bend upon temperature changes due to the differential expansion of the individual layers. Such a concept has been extensively exploited to produce micro-scale multilayered objects with self-folding properties using mostly photolithographic techniques.8–11
Self-shaping synthetic systems that rely on intrinsic molecular/atomic phase transitions or that are assembled from components exhibiting on-demand differential response have been explored or envisaged in numerous applications2 including biomedical devices,7 deployable space structures,12 drug delivery systems,13 morphing wings,14 smart textiles15 and self-healing composites.16 Despite the growing interest and enticing potential technologies, current self-shaping materials still suffer from major drawbacks. Because they rely on specific phase transitions at the atomic/molecular scale, shape-memory synthetic materials are sometimes only available in a narrow set of chemical compositions. Limited chemistries usually restrict the possible additional functionalities that can be readily and cost-effectively achieved, such as biocompatibility, bioresorbability, optical transparency and chemical inertness. Multi-component systems can potentially be built from a wider variety of chemical compositions, but typically require multiple photolithographic steps during processing and exhibit shape changes that are often limited to bending movements.
In contrast to synthetic systems, some natural materials utilize remarkably different approaches to accomplish shape changes upon external stimuli.17–26 Examples of these natural self-shaping materials are abundant in plants, ranging from pinecones to seedpods to tendrils and wheat awn structures. Instead of molecular phase transitions or combinations of different materials, many biological materials program their internal microstructure by controlling the local positioning and orientation of stiff fibers. Such fiber microstructures result in differential swelling/shrinkage or growth of the surrounding organic matrix to generate mechanical deformations that give rise to well-defined macroscopic changes in shape.20,27 Understanding and replicating such natural design principles may provide new pathways to create synthetic materials with autonomous self-shaping capabilities. Since this type of natural shape changing effect does not arise from phase transitions at the atomic/molecular level, self-shaping bioinspired materials can be potentially made with a wider variety of chemistries by properly designing the organization of their constituent building blocks at the microscale. Programming the shape change within the material's microstructure may also enable unusual shape changes by combining, for example, bending and twisting motions that are not straightforward in multi-component systems produced by photolithography.
Here, we discuss recent examples of bioinspired synthetic materials that can undergo shape changes by programming their reinforcement architecture at the microscale following the general design principles found in several biological systems. We start with plant systems and bioinspired composites in which the orientation of reinforcing fibers is tuned to enable bending and/or twisting upon swelling of their organic matrix. Natural and synthetic systems that are able to curl during growth are then discussed to illustrate the use of local asymmetric reinforcement of matrices as a means to control shape in growing dynamic materials. Finally, tunable interactions similar to those found between reinforcing fibers that control the mechanics of echinoderm (sea cucumber) tissues are also shown to be an effective approach to program shape changes within the microstructure of fiber-reinforced synthetic composites. Unfortunately these examples do not represent an exhaustive list. Instead, they are chosen to illustrate the rich variety of shape changes that can be synthetically achieved through this bioinspired approach and its broad flexibility in terms of the chemistry of the essential constituents required to build self-shaping microstructures. We conclude with some current challenges and possible opportunities enabled by such a bioinspired route.
Self-shaping through controlled reinforcement orientation
Many natural systems utilize programmed cellulose microfibril (CMF) architectures to restrict swelling or growth in certain directions and, in this way, promote organized, macroscopic self-shaping.19 With such a mechanism, tree branches bend up towards the sky,28 wheat awns dig their way into soil,17 and pinecones and other seedpods cleverly open to release seeds under optimum conditions29,30 (Fig. 1). In these self-shaping examples, natural systems predominantly rely on the control over the local orientation of the reinforcement rather than on the intrinsic molecular/atomic scale properties of the plant constituent materials.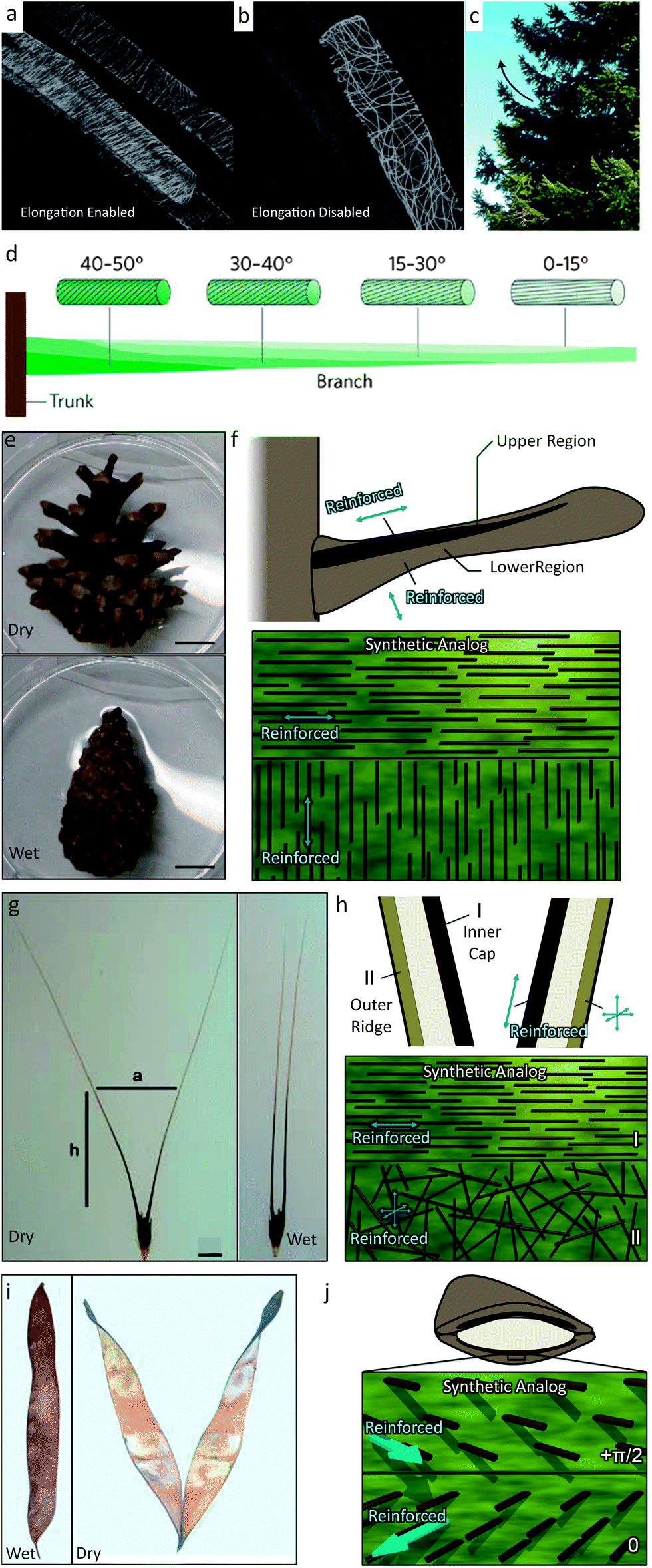 | ||
| Fig. 1 Biological systems that use fiber orientational control to enable structural bending and twisting include (a and b) the plant stem,31 (c and d) the tree branch,33 (e and f) the pine cone,25 (g and h) the wheat awn,18 and (i and j) the orchid tree seed pod.26 Images (a and b) are reproduced from Foster et al.31 with permission from Elsevier. Images (c and d) are reproduced from Fratzl and Barth33 with permission from Nature Publishing Group. Images (e–h and j) are reproduced from Erb et al.34 with permission from Nature Publishing Group. Image (g) is reproduced from Elbaum et al.32 with permission from AAAS. Image (i) is reproduced from Armon et al.26 with permission from AAAS. Scale bars in (e) and (g) are 1 cm. | ||
To this end, the orientation of CMFs is actively regulated in plant stems and tree branches. In certain plant stem systems, a random CMF orientation is displayed when the stem is programmed to restrict expansion in all directions.31 Instead, to promote 1-D expansion (growth), coordinated restructuring of CMF architectures is observed to align the reinforcement perpendicular to the stem (Fig. 1a and b). Tree branches influence their CMF orientation as a tree grows in response to applied stresses induced by gravity, wind or erosion. For example, the conifer pine branch transitions its CMF orientation along the length of a branch from low angles to higher angles. This heterogeneous architecture leads to the natural bending of the conifer tree branches to overcome gravity and regulate the stress in the branch28 (Fig. 1c and d). Active responses are necessarily complex requiring signal pathways between biomolecules to enable creation or redesign of the CMF architecture.
Elegant, active responses require live systems and are not very applicable for seed dispersal units, such as pinecones, wheat awns and seedpods, that need to undergo a CMF-mediated shape change while disconnected from their nutrient source. These seed dispersal units are instead passive systems that change shape in their ‘dying’ or dead state. In these examples, the CMF architecture therefore needs to be predetermined. Pinecones exhibit an elegant pre-programmed CMF architecture that allows them to remain closed when attached to the tree or under wet conditions and then to open and release seeds as they dry out.25 This architecture involves two unique regions of different predominant CMF alignments in each pinecone scale. The upper region of the scale consists of sclerenchyma fibers that have the predominant CMF direction along the length of the scale. The lower region consists of sclereids exhibiting a predominant CMF direction perpendicular to the plane of the scale (Fig. 1e and f). As the pinecone dries out, the lower region will contract due to water loss while the upper region is reinforced against lateral contraction. The two regions are tightly connected, representing a frustrated geometry and the entire scale will bend downward to minimize internal stresses.24 The wheat awn exhibits a similar predetermined architecture of two distinct reinforcement regions that leads to a macroscopic bending of the structure (Fig. 1g and h).32 The inner cap of the wheat awn is comprised predominantly of in-plane oriented CMFs while the outer ridge has a random CMF orientation. These different orientations lead to different preferred axes of expansion and contraction and represent another frustrated geometry. Upon dehydration, the wheat awn will bend its two stalks apart from each other. Another example is the seedpod of an orchid tree (Bauhinia variegata) (Fig. 1i and j).26 Each half of this seedpod is comprised of tissue that has an ordered CMF bilayer architecture. One layer has CMFs oriented at 0 radians while the other layer is oriented at +π/2 radians relative to the seedpod's long axis at +π/4 radians. As the seedpod is dehydrated, such as after falling from the orchid tree, the CMF architecture tends to contract along two orthogonal principle axes that are incompatible with one another. To maximize contraction along both axes the seedpod chirally twists as it dries. Because of their opposite CMF architectures, the two halves of the seedpod show different chirality, which allows the seeds to disperse. These passive systems enable dramatic macroscopic shape changes by simply predefining the CMF architecture in a swellable matrix.
Such passive systems have inspired new self-shaping strategies35 and can be recreated synthetically in manufactured composites by gaining control over fiber orientation during the assembly process. Commercial continuous-fiber reinforced composites have excellent fiber orientation and often exhibit structures similar to the pinecone architecture. During curing of these composites subtle volumetric changes that parallel pinecone dehydration have been shown to lead to the undesired bending of the structure.36 Instead, by making the volumetric response large, these shape change effects can be exaggerated to make composites that undergo significant geometric movements. Such a macroscopic shape change has been shown in synthetic composites by utilizing hydrogels (e.g. gelatin) that undergo dramatic volumetric growth upon hydration. To program the CMF architectures in hydrogel systems, one proven method is to make micron-sized reinforcing elements responsive to external magnetic fields by labeling them with superparamagnetic iron oxide nanoparticles.37 For the proper size of reinforcing particles, this labeling technique leads to an ultra-high magnetic response (UHMR) in which the reinforcing elements respond to very low magnetic fields in the order of 1 mT (Fig. 2a and b). Using this UHMR technique, 7.5 μm alumina microplatelets have been assembled in different architectures within hydrogels.34
 | ||
| Fig. 2 Bioinspired self-shaping synthetic systems with oriented reinforcement.34,37 (a and b) An ultra-high magnetic response (UHMR) has been observed (a) by attaching superparamagnetic iron oxide nanoparticles to the surface of reinforcing microparticles, which (b) enables the orientation of the microparticles under weak magnetic fields. (c) Gradually changing the orientation of reinforcement in a hydrogel similar to the CMF architecture in a conifer tree branch leads to (d) a local periodic curvature in a poly(vinyl alcohol)-based artificial system. Images (a–d) are reproduced from Erb et al.37 with permission from AAAS. (e–g) UHMR alumina microplatelets can also be positioned with magnetic fields in gelatin matrices to recreate CMF architectures of (e) pinecones, (f) wheat awns and (g) orchid tree seed pods. Hydrating and dehydrating these systems allow for macroscopic shape changes that mimic their natural counterparts. Images (e–g) are reproduced from Erb et al.34 with permission from Nature Publishing Group. Scale bars in (e), (f) and (g) are 1 cm. | ||
A reinforcement architecture similar to that of the conifer pine branch (Fig. 2c and d) has recently been replicated synthetically using poly(vinyl alcohol) matrices with embedded reinforcing UHMR microplatelets exhibiting gradually changing orientations.37 To achieve such architectures, an aqueous precursor suspension containing the polymer and microparticles was subjected to magnetic fields created from permanent magnets with ordered, linear magnetic domains. The resulting architecture (Fig. 2c) gradually bends upwards and downwards depending upon the local orientation of the reinforcing microplatelets (Fig. 2d). In addition to static magnets, rotating magnetic fields can also be utilized to enable complete orientation of the microplatelets to form the analogues of different CMF architectures.38 To recreate the pinecone structure, a gelatin bilayer was manufactured that exhibited in-plane oriented microplatelets in the top half and out-of-plane oriented microplatelets in the bottom half, as schematically shown in Fig. 1f. When these structures are cycled between dehydrated and hydrated states, they bend downward and upward, respectively, in a similar manner to the natural pinecone (Fig. 2e). Likewise, the natural wheat awn architecture could be recreated by producing a gelatin bilayer with in-plane oriented micro-reinforcement in the top half and randomized micro-reinforcement in the bottom half (Fig. 2f). The randomized reinforcement is achieved simply by not applying a magnetic field during suspension casting. Because of its slightly higher lateral swellability as compared to the top-layer, the randomly reinforced layer drives the bilayer again to bend. In addition to bending, the chiral twisting of the orchid tree seed pod could also be recreated by building platelet-reinforced gelatin bilayers that exhibit orthogonal reinforcement of the two layers both angled at π/4 radians relative to the long-axis of the material. Such fiber architectures caused the hydrogels to undergo chiral twisting (Fig. 2g).
To make use of self-shaping manufactured composites, structure–function relationships need to be established to predict material responsiveness and thus create a programming code for the production of materials that undergo well-defined shape changes. For bilayer fiber reinforced composites that bend, analytical models have been developed to predict the change in the curvature, Δκ, as a function of geometrical and materials parameters.24,39 According to Mahadevan et al.,24 the curvature of bilayers whose shape change is driven by humidity variations can be calculated as follows:
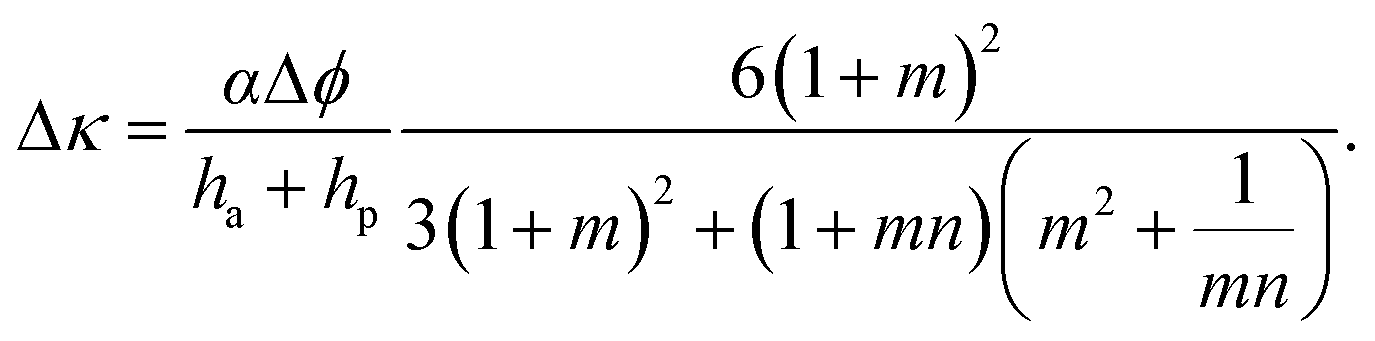 | (1) |
This model was applied to the pinecone system and was found to qualitatively follow the shape changing response of individual pinecone scales.24 To characterize the pinecone-inspired gelatin composites, several composites with varying active and passive layer heights were produced, swelled, and studied by Erb et al.34 The resulting structures led to a variety of curvatures with a maximum curvature when the passive layer is half the size of the active layer, which is in agreement with predictions given by eqn (1) (Fig. 3a and b). For the case of the orchid tree seed pod, the architecture used in the synthetic analogues exhibits a wide-range of self-shaping responses depending upon the angle θ of the reinforcing microstructure relative to the long-axis of the material (Fig. 3a and c–h). The normalized radius of curvature (![[r with combining tilde]](https://www.rsc.org/images/entities/i_char_0072_0303.gif) ) and normalized pitch (
) and normalized pitch (![[p with combining tilde]](https://www.rsc.org/images/entities/i_char_0070_0303.gif) of the resulting structures were found to conform to predictive energetic models of these geometrically frustrated structures developed by Armon and co-workers30 (lines in Fig. 3i).
of the resulting structures were found to conform to predictive energetic models of these geometrically frustrated structures developed by Armon and co-workers30 (lines in Fig. 3i).
 | ||
| Fig. 3 Structure–function relationships in self-shaping synthetic systems with oriented reinforcement.34 (a) Definition of the orientation angle, θ, of orthogonal bilayer reinforcement architectures that produce bending and twisting. (b) Dependence of the curvature, Δκ, of pinecone-inspired gelatin composites on the layer thickness ratio, m, for structures undergoing pure bending. This curvature is fit using eqn (1) (lines) for composites of different active and passive layer height ratios (m). (c–h) Gelatin composites inspired by orchid tree seed pods exhibit shape changes from pure twisting (c and f) to pure bending (e and h) depending upon the orientation of the reinforcing elements, θ, with respect to the long-axis of the material. (i) Dependence of the normalized radius of curvature, r, and pitch, p, as a function of the orientation angle, θ, for structures undergoing a wide range of shape changes from pure bending (θ = 0 and ±π/2) to pure twisting (θ = ± π/4). Reproduced from Erb et al.34 with permission from Nature Publishing Group. All scale bars are 0.5 cm. | ||
Self-shaping through controlled reinforcement distribution
Chiral twisting is ubiquitous in the natural world. Not only do the orchid tree seed pods chirally twist, but also DNA molecules self-assemble into helices, the coiled shells of gastropods chirally twist during growth, and climbing plants coil tendrils around supports in near-perfect helices.40 While in molecular systems, such as DNA, twisting arises from the chiral nature of the constituent single strands, there is no evidence that macroscale chirality is driven by nanoscale chiral precursors in many twisting plant systems. Instead, the macroscale chirality is oftentimes just a result of fiber architectures at the microscale, which lead to frustrated geometries that are driven to twist into other configurations exhibiting minimum energy. The coiling tendril of a cucumber plant is an illustrative example of macroscale chirality in which the minimum energy twisted geometry is programmed within the microstructure of the material by controlling the distribution of reinforcing elements in a continuous matrix (Fig. 4a). | ||
| Fig. 4 Tendrils of climbing plants. (a) The tendril of a cucumber plant twists during growth after making contact with a support. This twisting appears to be a result of a tissue morphosis that creates a stiff, lignified fiber ribbon with asymmetric reinforcement distribution along one side of the tendril (shown in a1 and a2). Reproduced from Gerbode et al.41 with permission from AAAS. Scale bars: 100 μm in (a1) and 10 μm in (a2). (b) Unbound tendrils are known to form curly q's. Reproduced from Godinho et al.43 with permission from the Royal Society of Chemistry. (c) Tendrils that are initially bound between two points prior to twisting must maintain zero net twist and develop perversions (region enclosed by the circle). Reproduced from Godinho et al.42 with permission from the Royal Society of Chemistry. | ||
During its growth, a tendril of the cucumber plant first extends itself straight until it contacts and attaches to a support.40 Upon contact, it has been proposed41 that one side of the coil exhibiting a ribbon of gelatinous fiber cells undergoes tissue morphosis in the form of lignification, water removal, and development of oriented CMFs (region highlighted in the insets of Fig. 4a). The asymmetric distribution of reinforcement (e.g. lignin) within such a fiber ribbon creates a frustrated geometry arising from the differential local growth of the cell layers with different reinforcement levels. As the tendril morphs and grows, the minimal energy solution is for the tendril to chirally twist to minimize the length of the more reinforced portion of the ribbon while maximizing the length of the less reinforced side.41 Sometimes this twisting can begin with the tendril not contacting a point of support, which results in a coil (Fig. 4b). However, if the twisting conformation initiates only after the tendril contacts an opposite point of support, there must be zero net twist in the whole length of the tendril. To accommodate this fixed boundary condition, the tendril develops a perversion that is a point of helix reversal where the tendril changes from left-handed to right-handed chirality42 (Fig. 4c). If these boundary points are then compressed, the radius of curvature of the twists increases. For the case where the boundary points extend, it is intuitively expected that the radius of curvature will decrease and the twists will unwind themselves. Surprisingly, extension of the boundary points may actually lead to over-winding of tendrils exhibiting a perversion. In this case, over-winding occurs to preserve the equilibrium radius of curvature of the tendril during extension, which is made possible due to the ability of the perversion to rotate freely.41 Understanding the rich mechanisms that drive chiral twisting in plant systems may allow us to reapply such microstructural designs to drive powerful material shape changes in synthetic analogues.
Synthetic systems that exhibit identical cylindrical twisting to the cucumber tendril have been reported. Canejo and co-workers created materials with a chiral fiber architecture by electrospinning acetoxypropylcellulose (APC) fibers at high volume fractions in an anhydrous dimethylacetamide (DMAc) solvent.44 At high enough volume fractions, the APC self-assembles into a liquid crystalline cholesteric phase43 and the solution is sufficiently fluid to be pumped through a capillary for electrospinning (Fig. 5a). In the absence of shear, the APC fibers arrange in a chiral configuration with the director axis normal to the capillary wall and a disclination line that coincides with the capillary axis (Fig. 5b).45 As this liquid crystalline solution is forced through the capillary during the electrospinning process (Fig. 5a), the APC solution is subjected to a parabolic flow, which leads to gradually higher shear stresses as one moves from the center to the walls of the capillary. This causes the disclination line to shift from the central axis to the edge of the capillary, ultimately forming a chiral topological defect along the capillary (Fig. 5b). At high enough flow rates, the disclination line is sufficiently displaced to run chirally next to the capillary wall. This disclination line forms a ‘hard’ helix that is eventually detectable in the spun macro-fiber, as evidenced by the magnetic resonance images shown in Fig. 5b. The ‘hard’ disclination line parallels the stiffened fibrous section of the cucumber plant tendril and acts as the guiding microstructural element that leads to a shape change. The asymmetric reinforcement in the synthetic system occurs between the disclination line and the cholesteric APC bulk phase (Fig. 5b), whereas for the cucumber tendril such asymmetry seems to be restricted to the fiber ribbon itself (Fig. 4a). Likely, as the solvent is evaporated from the APC solution during spinning and drying, the fiber undergoes a shrinking that is preferentially reduced along the ‘hard’ disclination line. This microstructure produces a frustrated geometry that responds by twisting to minimize the system's energy. Thus, chirality in the macroscopic material is programmed using shear fields to generate a self-shaping microstructure that is similar to the reinforcement distribution of twisting climbing tendrils (Fig. 5c and d). This is an interesting example in which the topological defect arising from the constrained packing of chiral molecules rather than the chirality of the molecule itself is responsible for the shape change.
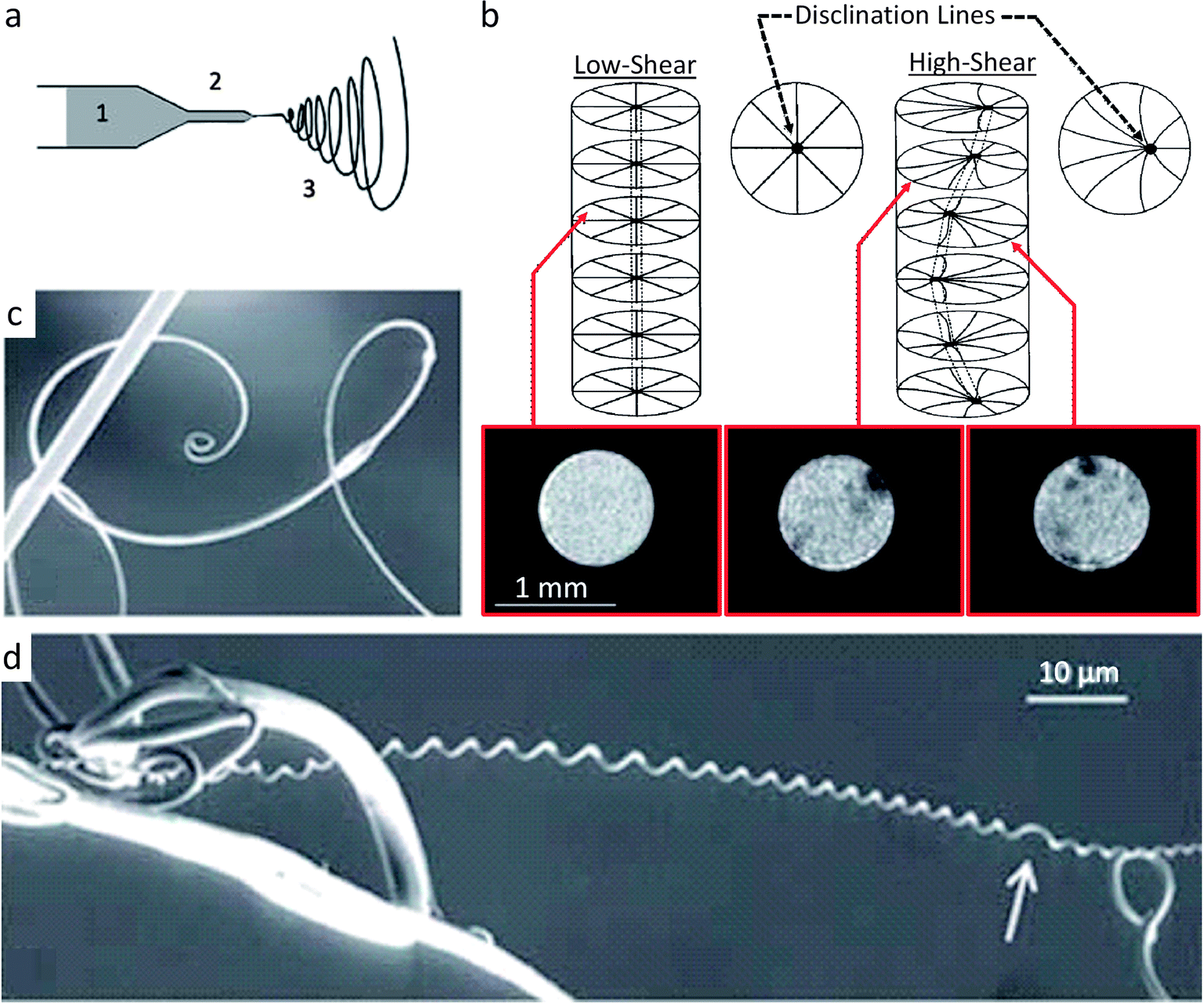 | ||
| Fig. 5 Synthetic route for making fibers that mimic the twisting shape change found in climbing plant tendrils.43 (a) Electrospinning of a solution of acetoxypropylcellulose (1) (APC) through a capillary (2) to generate a twisting macroscopic fiber (3). (b) Disclination line which parallels the stiff fiber of the cucumber plant tendril is seen as the dark (hard) regions in the inset MRI cross-sectional scans of the APC capillary flow. (c and d) Upon electrospinning at high shear, the macro-fibers twist to reduce the high elastic energy created by the disclination line. Similar to plant tendrils, APC macro-fibers connected only at one point can form spirals (c), while APC macro-fibers that are connected at two points form helical twists (d) often with perversions (white arrow). Images (a, c and d) are reproduced from Godinho et al.43 with permission from the Royal Society of Chemistry. Image (b) is adapted from Godinho et al.43 and Cronin et al.45 with permission from the Royal Society of Chemistry and Taylor & Francis. | ||
Canejo et al. also worked to build a programming code for the shape change exhibited by these microstructures.44 By analyzing the geometry of fibers generated under different spinning conditions, a linear relationship was found between the fiber diameter and the chiral pitch (Fig. 6). Such a correlation resembles the effect of the thickness and width on the geometry of twisting ribbons, whose final shape is determined by a competition between bending and stretching elastic energies.26 For ribbons, higher pitches are obtained when the system is dominated by the stretching energy. Although a more thorough analysis is required, this comparison suggests that larger APC fiber diameters result in higher pitches because thicker fibers dissipate more energy by stretching than by bending.44
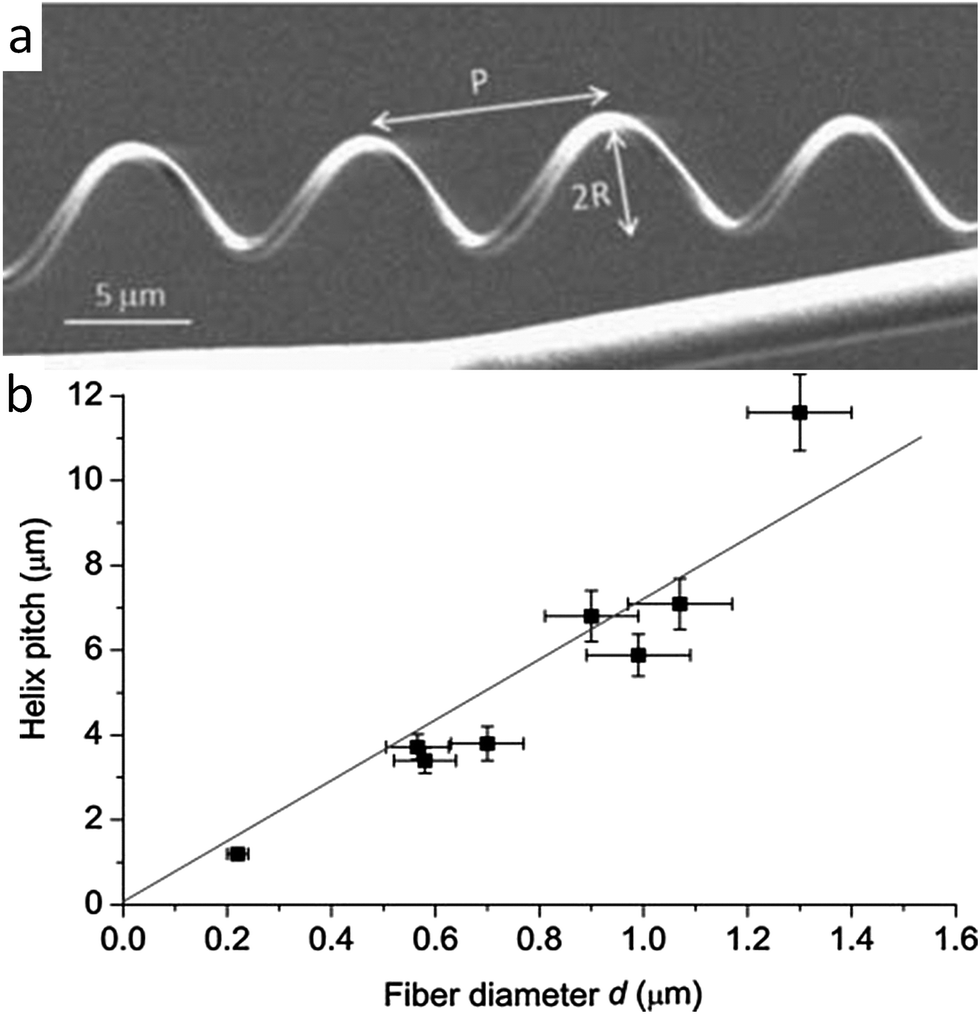 | ||
| Fig. 6 Geometrical features of fibers that mimic the twisting shape change found in climbing plant tendrils. (a) Synthetic chiral fibers produced by electrospinning, highlighting the pitch P and the radius R of the generated helical geometry. Reproduced from Godinho et al.43 with permission from the Royal Society of Chemistry. (b) Linear relationship found between the helical pitch and the diameter of the macrofiber. Reproduced from Canejo et al.44 with permission from Wiley. | ||
Self-shaping via switchable reinforcement interactions
In addition to the orientation and spatial distribution of reinforcing elements, many biological materials also exhibit active control over the interactions and cross-links between reinforcing fibers and filaments. As a general illustrative example, dynamic control over the cross-linking of filamentous proteins in the cytoskeleton enables shape changes, division and coordinated motion of living cells.46,47 Controlled interactions between structural fibers may also occur in the extracellular matrix around cells. A well-known example is the ability of echinoderms to reversibly switch the interactions between reinforcing collagen fibers to quickly change the mechanics of their dermal connective tissue.48 Using this mechanism, the dermal tissue of sea cucumbers can undergo a 10-fold increase in its elastic modulus within a timeframe of only a few seconds (Fig. 7). Although in echinoderms it provides a defensive function rather than a shape changing mechanism, the possibility of reversibly switching the interactions between reinforcing fibers has recently been exploited as a simple approach to generate self-shaping synthetic materials through microstructural design.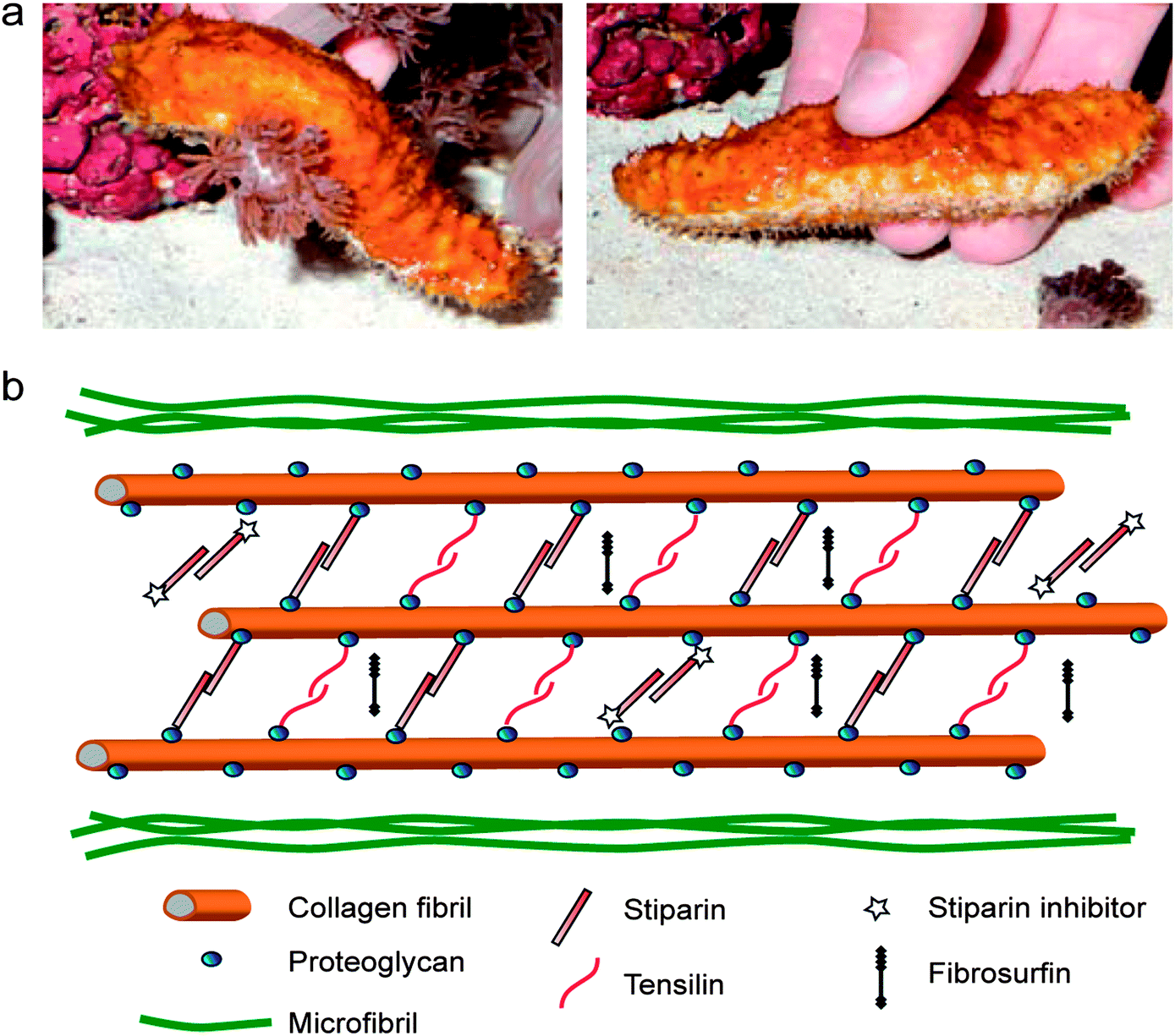 | ||
| Fig. 7 The stiffening response of sea cucumbers triggered by chemical stimuli. (a) A sea cucumber in its soft (left) and stiff (right) states. Reproduced from Capadona et al.52 with permission from AAAS. (b) Hypothetical architecture of collagen fibers and other biomolecules in the dermal tissue of sea cucumbers, highlighting the reversible cross-linking molecules presumably involved in the stiffening process. Adapted from Wilkie48 with permission from Springer. | ||
Systematic studies have shown that the reversible tuning of fiber interactions in the dermal tissue of sea cucumbers is promoted by chemicals that are released from cells present in the outer and inner dermis of the animal.49–51 The glycoprotein stiparin has been suggested to play a key role in the aggregation of collagen fibers through the formation of weak bonds with proteoglycans present on the surface of adjacent reinforcing fibrils (Fig. 7). This leads to the rapid formation of a percolating network of collagen fibrils that significantly increases the elastic modulus of the dermal tissue. However, because of its weak nature, such cross-linking bonds can be disrupted in the presence of another protein, a stiparin-inhibitor, to eventually result in softening of the tissue. Other molecules have been suggested to participate in this stiffening/softening mechanism, but their exact roles are still the subject of investigation (Fig. 7).
The concept of switchable interactions between reinforcing building blocks was replicated artificially using cellulose nano-whiskers as reinforcing elements in combination with different polymeric matrices.52,53 In these systems, the nano-whiskers interact through hydrogen bonding to form a stiff percolating network throughout the composite. Water molecules are then used as chemical triggers to soften the material via the formation of competitive hydrogen bonds that disrupt the initial percolating network. Switching on and off the interactions between reinforcing whiskers can change the elastic modulus of the composite by as much as one order of magnitude if the volume fraction of reinforcement is sufficiently high to ensure percolation of the fibers (Fig. 8). The fact that the elastic modulus of water-free samples can be reasonably described by a percolation model (Fig. 8b) confirms that the stiffening effect is indeed a result of the onset of cross-linking interactions between the cellulose nanowhiskers. Rupture of such cross-links through water-induced competitive hydrogen bonds reduces the elastic modulus to values close to the level expected from the Halpin–Kardos model for non-interacting whiskers (Fig. 8b).
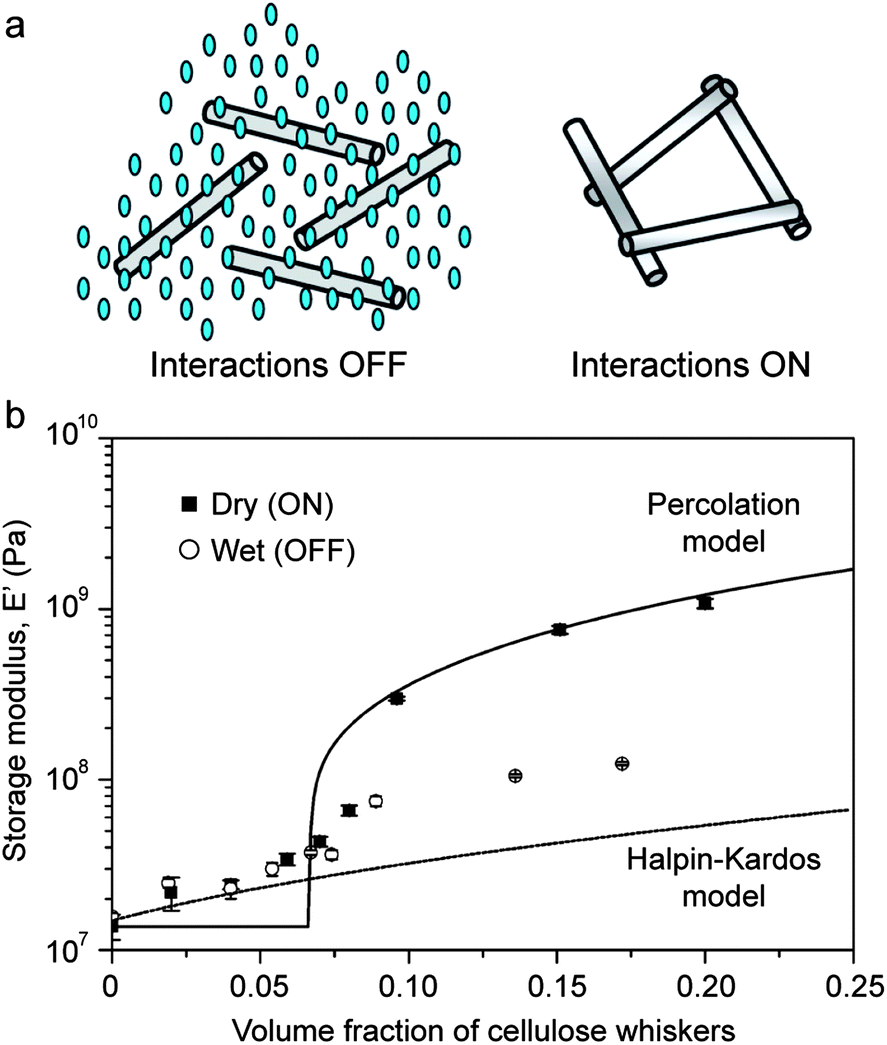 | ||
| Fig. 8 Reversible stiffening/softening of bioinspired composites through tunable reinforcement interactions. (a) Schematics showing the lack of fiber interactions in the presence of water (left) and the stiff percolating network of fibers generated in the dry state (right). Reproduced from Capadona et al.52 with permission from AAAS. (b) Effect of the type of fiber interactions on the elastic modulus of polyurethane reinforced with cellulose nanowhiskers.54 Reproduced from Mendez et al.54 with permission from the American Chemical Society. | ||
Inspired by the unique chemomechanical response of sea cucumbers, the reversible formation of an elastic network of percolating fibers has recently been exploited to create composites exhibiting shape-memory effects that are triggered by a chemical stimulus. In this case, the ability to form a strong percolating network of reinforcing fibers was used to temporarily store a large amount of elastic energy that is initially applied to the surrounding polymer matrix of the self-shaping material. On-demand rupture of the stiff percolating network using a chemical trigger eventually releases the elastic energy stored within the system to cause a significant shape change of the entire composite (Fig. 9a).
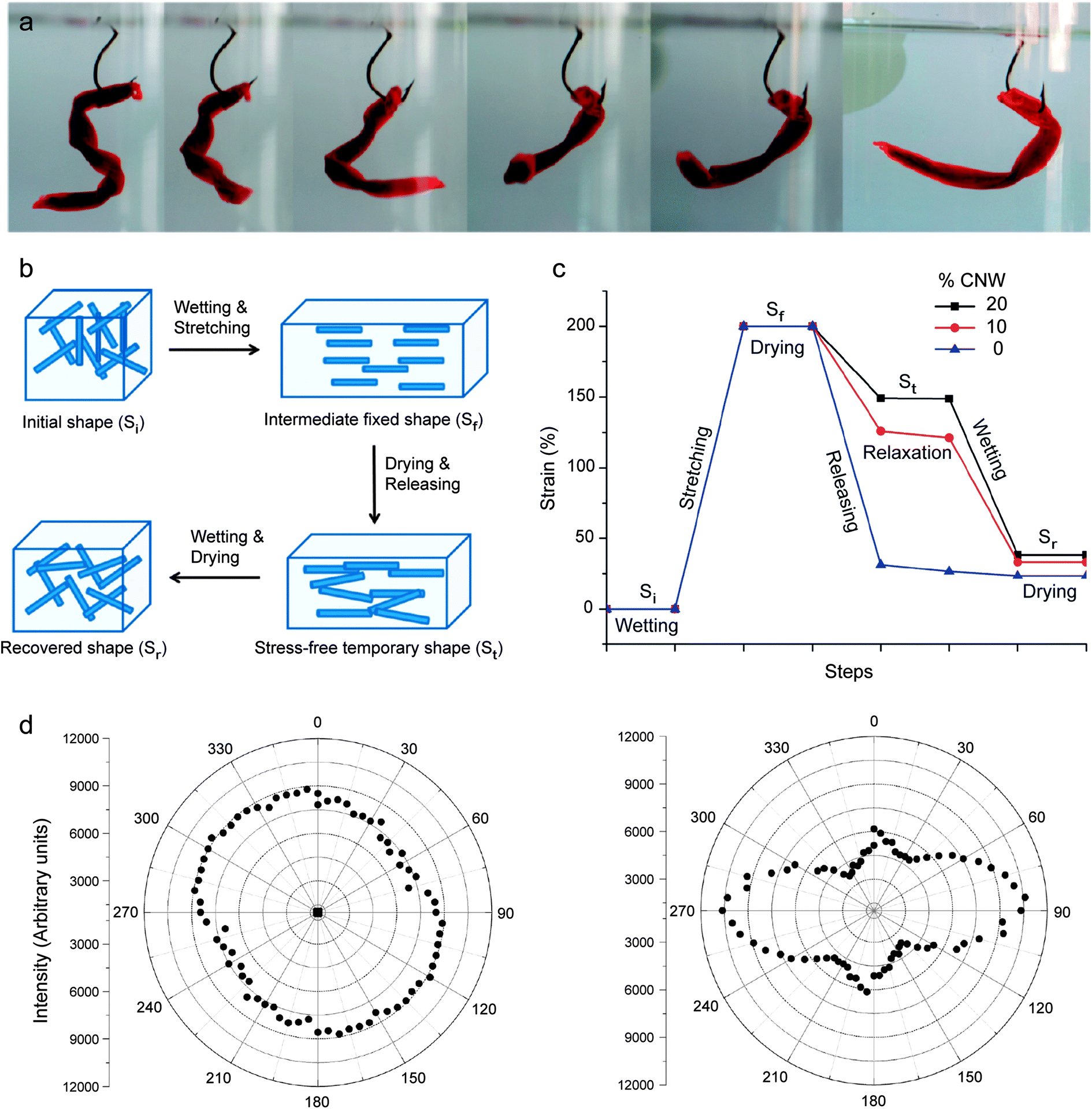 | ||
| Fig. 9 Synthetic composites that undergo reversible shape changes through switchable interactions between reinforcing fibers. (a) “Fish lure” synthetic composite that self-shapes upon water uptake. (b) Schematics illustrating the steps required to fix a temporary shape upon drying and to recover the original shape upon wetting. (c) Mechanical strains applied and recovered along the shape changing process of polyurethanes containing 0, 10 and 20% cellulose nanowhiskers (CNWs). (d) Polar plots based on polarized Raman spectroscopy showing the random orientation (left) and the preferential alignment (right) of nanowhiskers in the undeformed and uniaxially stretched composites containing 20% CNW, respectively.54 Reproduced from Mendez et al.54 with permission from the American Chemical Society. | ||
The steps required to obtain the initial temporary shape and to eventually change it into the final stress-free state are schematically illustrated in Fig. 9b and c for composites consisting of a polyurethane matrix reinforced with cellulose nanowhiskers. In this illustrative example, the composite is first exposed to a wet environment to switch off the interactions between the reinforcing whiskers. Because of the strong elastic nature of the polyurethane matrix, the absence of cross-linking between cellulose whiskers enables uniaxial stretching of the composite up to 200% strain. Such high stretchability also leads to the alignment of whiskers in the stretching direction, as schematically illustrated in Fig. 9b.
Removal of water from the composite while still in the stretched state leads to the formation of a stiff percolating network of cellulose whiskers that are cross-linked through hydrogen bonding. When the applied stress is released after complete drying, the stiff cellulose network is able to retain a large fraction of the initial mechanical deformation imposed during stretching. Thus, a consolidated temporary shape (St) is obtained. Polarized Raman spectroscopy of composites before and after stretching and drying shows that the high degree of alignment of whiskers achieved during the initial stretching step is still observed in the temporarily shaped samples (Fig. 9d). This supports the idea that the temporary shape is maintained by the formation of cross-linking interactions between whiskers, which do not let the elastomeric matrix relax back to its original unstretched configuration.
Switching the composite from its temporary shape to the more energetically favorable unstressed shape (Sr) can be easily triggered by exposing the material to water. Competitive hydrogen bonding of the water molecules between adjacent cellulose whiskers eventually breaks the stiff network again to enable full relaxation of the initially stretched polyurethane matrix. The entire cycle from the initial mechanical deformation of the composite into a temporary shape followed by its full relaxation in the presence of water molecules can be repeated multiple times. Applying such concepts to advanced composites requires the utilization of reinforcing fibers whose interactions are sensitive to a specific external stimulus, as is the case of cellulose whiskers in the presence of water. Once this criterion is met, bulk composites can be mechanically shaped into any arbitrary geometry and eventually relaxed to their original shape using a specific stimulus as an external trigger (Fig. 9a).
Conclusions and outlook
Designing the reinforcement architecture of polymers and composites at the microscale is a powerful approach to create bioinspired materials that can self-shape into pre-programmed geometries during processing or stimulated volumetric changes. The examples highlighted here illustrate three bioinspired mechanisms that have been synthetically replicated to achieve tunable shape changes through microstructural design. These are (i) the controlled orientation of reinforcing building blocks to enable self-shaping upon swelling or growth of the composite; (ii) the asymmetric distribution of reinforcing fibers and macromolecules within a polymer-based material to promote self-shaping during its processing or growth; and (iii) the switchable interactions between reinforcing fibers to allow for on-demand formation and breakdown of a percolating elastic network able to consolidate composites into a temporary shape.By relying on the architecture of anisotropic building blocks at the microscale, these bioinspired mechanisms represent a new route to obtain self-shaping materials using basic constituents that would otherwise lack the molecular phase transitions typically needed to create shape-memory effects in polymers and metallic alloys. This flexibility with regard to the chemistry of the building blocks opens several new opportunities to achieve programmed, stimulated shape changes in artificial systems. For example, standard bioresorbable polymers and inorganic materials that are already approved by regulatory agencies can be combined into composite architectures exhibiting self-shaping features using some of the techniques discussed above. Other functionalities, such as tunable magnetic or optical properties, can also be easily incorporated into these composites without impairing their primary shape-changing function. In fact, combining bioinspired self-shaping architectures at the microscale with phase transitions of polymers at the molecular scale should enable programming of multiple shape changes in one single composite. This approach can potentially lead to hierarchically structured composites exhibiting a multitude of shape-changing effects programmed at different length scales of the material. Following this approach, the number of temporary shapes that can be programmed in shape-memory polymers could be further extended beyond the number of discrete reversible phase transitions within the material.
As a new pathway to generate shape changes in synthetic materials, bioinspired routes also have to overcome a number of challenges before their implementation in real applications. For example, self-shaping composites triggered by water uptake remain significantly weaker than their biological counterparts. A deeper understanding of the self-shaping mechanisms in biological and bioinspired materials is also required to enable the development of robust models to predict the dynamics and final geometries of such shape-changing systems. Moreover, a rich variety of other shape-changing materials existing in nature are yet to be thoroughly investigated. As our knowledge about the self-shaping mechanisms in natural materials advances, new processing challenges will have to be addressed to implement synthetically the intricate microstructure of shape-changing biological systems. Finally, combining the elaborate microstructural design of biological materials with the wealth of chemical compositions available in synthetic systems can potentially enable the creation of multifunctional self-shaping materials whose functionalities and properties can even surpass those of the biological systems taken as the original sources of inspiration.
References
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- J. S. Leng, X. Lan, Y. J. Liu and S. Y. Du, Prog. Mater. Sci., 2011, 56, 1077–1135 CrossRef CAS PubMed.
- Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS PubMed.
- S. V. Ahir and E. M. Terentjev, Nat. Mater., 2005, 4, 491–495 CrossRef CAS PubMed.
- C. L. van Oosten, C. W. M. Bastiaansen and D. J. Broer, Nat. Mater., 2009, 8, 677–682 CrossRef CAS PubMed.
- K. Otsuka and C. M. Wayman, Shape Memory Materials, Cambridge University Press, Cambridge, UK, 1999 Search PubMed.
- A. Lendlein and R. Langer, Science, 2002, 296, 1673–1676 CrossRef PubMed.
- D. H. Gracias, V. Kavthekar, J. C. Love, K. E. Paul and G. M. Whitesides, Adv. Mater., 2002, 14, 235–238 CrossRef CAS.
- E. Smela, O. Inganas and I. Lundstrom, Science, 1995, 268, 1735–1738 CAS.
- L. Ionov, Soft Matter, 2011, 7, 6786–6791 RSC.
- E. Hawkes, B. An, N. M. Benbernou, H. Tanaka, S. Kim, E. D. Demaine, D. Rus and R. J. Wood, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12441–12445 CrossRef CAS PubMed.
- K. Gall, M. Mikulas, N. A. Munshi, F. Beavers and M. Tupper, J. Intell. Mater. Syst. Struct., 2000, 11, 877–886 CAS.
- H. Y. He, J. J. Guan and J. L. Lee, J. Controlled Release, 2006, 110, 339–346 CrossRef CAS PubMed.
- J. L. Reed, C. D. Hemmelgarn, B. M. Pelley and E. Havens, in Smart Structures and Materials, ed. E. V. White, Industrial and Commercial Applications of Smart Structures Technologies, 2005, vol. 5762, pp. 132–142 Search PubMed.
- H. Tobushi, H. Hara, E. Yamada and S. Hayashi, Smart Mater. Struct., 1996, 5, 483–491 CrossRef CAS.
- M. Thomas, B. Thomas, H. David and H. Chistopher, in 49th AIAA/ASME/ASCE/AHS/ASC Structures, Structural Dynamics, and Materials Conference, American Institute of Aeronautics and Astronautics, 2008 Search PubMed.
- R. Elbaum, L. Zaltzman, I. Burgert and P. Fratzl, Science, 2007, 316, 884–886 CrossRef CAS PubMed.
- P. Fratzl, R. Elbaum and I. Burgert, Faraday Discuss., 2008, 139, 275–282 RSC.
- I. Burgert and P. Fratzl, Philos. Trans. R. Soc., A, 2009, 367, 1541–1557 CrossRef CAS PubMed.
- M. J. Harrington, K. Razghandi, F. Ditsch, L. Guiducci, M. Rueggeberg, J. W. C. Dunlop, P. Fratzl, C. Neinhuis and I. Burgert, Nat. Commun., 2011, 2, 337 CrossRef PubMed.
- J. Braam, New Phytol., 2005, 165, 373–389 CrossRef PubMed.
- O. Vincent, C. Weisskopf, S. Poppinga, T. Masselter, T. Speck, M. Joyeux, C. Quilliet and P. Marmottant, Proc. R. Soc. B, 2011, 278, 2909–2914 CrossRef PubMed.
- S. Poppinga, S. R. H. Hartmeyer, R. Seidel, T. Masselter, I. Hartmeyer and T. Speck, PLoS One, 2012, 7, e45735 CAS.
- E. Reyssat and L. Mahadevan, J. R. Soc., Interface, 2009, 6, 951–957 CrossRef CAS PubMed.
- C. Dawson, J. F. V. Vincent and A.-M. Rocca, Nature, 1997, 290, 668 CrossRef PubMed.
- S. Armon, E. Efrati, R. Kupferman and E. Sharon, Science, 2011, 333, 1726–1730 CrossRef CAS PubMed.
- Y. Forterre, J. M. Skotheim, J. Dumais and L. Mahadevan, Nature, 2005, 433, 421–425 CrossRef CAS PubMed.
- P. Fratzl and F. G. Barth, Nature, 2009, 462, 442–448 CrossRef CAS PubMed.
- C. Dawson, J. F. V. Vincent and A.-M. Rocca, Nature, 1997, 390, 668 CrossRef CAS PubMed.
- S. Armon, E. Efrati, R. Kupferman and E. Sharon, Science, 2011, 333, 1726–1730 CrossRef CAS PubMed.
- R. Foster, O. Mattsson and J. Mundy, Trends Plant Sci., 2003, 8, 202–204 CrossRef CAS.
- R. Elbaum, L. Zaltzman, I. Burgert and P. Fratzl, Science, 2007, 316, 884–886 CrossRef CAS PubMed.
- P. Fratzl and F. G. Barth, Nature, 2009, 462, 442–448 CrossRef CAS PubMed.
- R. M. Erb, J. S. Sander, R. Grisch and A. R. Studart, Nat. Commun., 2013, 4, 1712 CrossRef PubMed.
- Z. L. Wu, M. Moshe, J. Greener, H. Therien-Aubin, Z. H. Nie, E. Sharon and E. Kumacheva, Nat. Commun., 2013, 4, 1586 CrossRef PubMed.
- D. W. Radford, J. Compos. Technol. Res., 1993, 15, 290–296 CrossRef CAS.
- R. M. Erb, R. Libanori, N. Rothfuchs and A. R. Studart, Science, 2012, 335, 199–204 CrossRef CAS PubMed.
- R. M. Erb, J. Segmehl, M. Charilaou, J. F. Loffler and A. R. Studart, Soft Matter, 2012, 8, 7604–7609 RSC.
- S. Timoshenko, J. Opt. Soc. Am. Rev. Sci. Instrum., 1925, 11, 233–255 CrossRef CAS.
- C. Darwin, J. Linn. Soc. London, Bot., 1867, 9, 1–118 CrossRef.
- S. J. Gerbode, J. R. Puzey, A. G. McCormick and L. Mahadevan, Science, 2012, 337, 1087–1091 CrossRef CAS PubMed.
- M. H. Godinho, J. P. Canejo, L. F. V. Pinto, J. P. Borges and P. I. C. Teixeira, Soft Matter, 2009, 5, 2772–2776 RSC.
- M. H. Godinho, J. P. Canejo, G. Feio and E. M. Terentjev, Soft Matter, 2010, 6, 5965–5970 RSC.
- J. P. Canejo, J. P. Borges, M. H. Godinho, P. Brogueira, P. I. C. Teixeira and E. M. Terentjev, Adv. Mater., 2008, 20, 4821–4825 CrossRef CAS.
- D. W. Cronin, E. M. Terentjev, R. A. Sones and R. G. Petschek, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1994, 238, 167–177 CrossRef CAS.
- B. Wagner, R. Tharmann, I. Haase, M. Fischer and A. R. Bausch, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13974–13978 CrossRef CAS PubMed.
- M. L. Gardel, K. E. Kasza, C. P. Brangwynne, J. Liu and D. A. Weitz, in Methods in Cell Biology, Elsevier, 2008, vol. 89 Search PubMed.
- I. C. Wilkie, in Echinodermata, ed. V. Matranga, Springer Berlin Heidelberg, 2005, vol. 39, ch. 10, pp. 221–250 Search PubMed.
- J. A. Trotter, G. Lyons-Levy, K. Chino, T. J. Koob, D. R. Keene and M. A. L. Atkinson, Matrix Biol., 1999, 18, 569–578 CrossRef CAS.
- J. A. Trotter, G. Lyons-Levy, D. Luna, T. J. Koob, D. R. Keene and M. A. L. Atkinson, Matrix Biol., 1996, 15, 99–110 CrossRef CAS.
- G. K. Szulgit and R. E. Shadwick, J. Exp. Biol., 2000, 203, 1539–1550 CAS.
- J. R. Capadona, K. Shanmuganathan, D. J. Tyler, S. J. Rowan and C. Weder, Science, 2008, 319, 1370–1374 CrossRef CAS PubMed.
- K. Shanmuganathan, J. R. Capadona, S. J. Rowan and C. Weder, Prog. Polym. Sci., 2010, 35, 212–222 CrossRef CAS PubMed.
- J. Mendez, P. K. Annamalai, S. J. Eichhorn, R. Rusli, S. J. Rowan, E. J. Foster and C. Weder, Macromolecules, 2011, 44, 6827–6835 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |