 Open Access Article
Open Access ArticleNonlamellar lipid liquid crystalline model surfaces for biofunctional studies†
Maria
Wadsäter
*a,
Justas
Barauskas
bc,
Tommy
Nylander
a and
Fredrik
Tiberg
ab
aDepartment of Physical Chemistry, Lund University, P.O. Box 124, SE-22100, Lund, Sweden. E-mail: maria.wadsater@fkem1.lu.se
bCamurus AB, Ideon Science Park, Gamma Building, Sölvegatan 41, SE-22379 Lund, Sweden. E-mail: info@camurus.com
cBiomedical Science, Faculty of Health and Society, Malmö University, SE-20506 Malmö, Sweden. E-mail: justas.barauskas@mah.se
First published on 7th August 2013
Abstract
Nonlamellar lipid liquid crystalline (LC) bulk phases and dispersions show promise as functional nanostructured materials for potential use as controlled release matrices e.g. in pharmaceuticals. Herein, methods for preparing and characterizing thin films of lipid liquid crystalline phases on solid surfaces are presented. The thickness, hydration phase structure and surface topography of spin-coated films of mixtures of soy phosphatidylcholine and glycerol dioleate are characterized by means of spectroscopic ellipsometry, small angle X-ray diffraction and atomic force microscopy. Besides being useful as bioadhesive drug delivery systems, the lipid nonlamellar LC films produced may also be exploited as model surfaces for studying properties such as bioadhesion and biodegradation.
Lyotropic nonlamellar lipid liquid crystalline (LC) phases, such as cubic, hexagonal and sponge phases, have been recognized as potential drug delivery and food nutrient carrier systems for several decades.1–3 The three-dimensional nano-structured framework of interconnected or discrete lipid and aqueous domains allows for solubilisation of both hydrophobic and hydrophilic substances as well as amphiphilic molecules, such as peptides and small proteins.4,5 Besides preventing rapid in vivo biodegradation of sensitive pharmaceutical substances, the LC structure can be designed to effectively restrict the diffusive transport between the space divided nano-domains, providing extended drug release profiles, largely controlled by biodegradation.6 Reversed hexagonal (H2) and cubic micellar (I2) phases have been found to be of particular interest for sustained release drug delivery applications, where active pharmaceutical ingredients are encapsulated in the lipid nanostructure, featuring discrete aqueous domains.7,8 Furthermore, the H2 and I2 phases are stable in excess of water and can be dispersed into nanometer-sized colloidal particles, i.e. hexosomes and cubosomes.9 Thus, the nonlamellar liquid crystalline bulk phases and dispersions offer promising and versatile drug delivery systems also in terms of the route of administration. For instance, the I2 phase was proposed to be able to serve as an in vivo drug reservoir10 and the liquid crystalline nanoparticles as injectable drug carriers.11 The I2 phase of soy phosphatidylcholine (SPC) and glycerol dioleate (GDO) has also been demonstrated to have excellent mucoadhesive film properties with an optimum lipid ratio of about 35/65 wt%/wt% SPC/GDO. The mucoadhesive and protective properties of this lipid mixture are exploited in episil®, a medical product for local treatment of pain in patients with e.g. oral mucositis, a painful and potentially treatment limiting side effect of chemotherapy and radiotherapy in cancer patients.12
Various polar lipid and surfactant systems form reversed-phase nonlamellar liquid crystalline structures.13,14 However, for drug carrier applications the system is ideally comprised of a mixture of a lamellar (Lα) phase forming lipid and a lipid that forms structures with high negative curvature. Undesired phase transitions can thus be counteracted by fine-tuning the lipid ratio. Mixtures of GDO and SPC are known to form a variety of different reversed LC phases. With increasing fractions of GDO, H2, I2 and L2 and in some cases several intermediate phases are observed in excess of water.15 Liquid crystalline phases of SPC and GDO have shown advantageous properties e.g. slow drug release profiles and low haemolytic activity, compared to other lipid systems.8,16 Furthermore, the uncomplicated phase behaviour of e.g. 35/65 wt%/wt% SPC/GDO upon hydration, which directly forms a single cubic micellar (Fd3m)17–19 phase from the L2 phase,20 is practically advantageous in many applications.
This communication concerns the controllable formation and characterization of thin nonlamellar liquid crystalline lipid films on a solid support. Such films can be useful as model surfaces with tuneable properties for routine studies of e.g. bioadhesion and biodegradation phenomena of the nonlamellar LC phase. Interfacial vs. interior LC phase properties can be investigated and a number of additional analysis methods become available through surface sensitive techniques. For instance, the supported lipid bilayer is a typical example of a well-characterized biomimetic model of the biological cell membrane,21 frequently used to study membrane interactions.22 Fusion of lipid vesicles and transfer of lipid monolayers from the air–liquid interface onto a solid surface are standard procedures frequently used to prepare these lipid films.23,24 In spite of the importance of nonlamellar phases in many biological systems,25 so far only few studies of nonlamellar lipid-aqueous layers formed by adsorption of discrete nonlamellar particles have been reported.26,27
In this study we use spin-coating as an alternative technique to prepare thin lipid films on solid surfaces.28 The formation of lipid bi- and multilayers upon hydration of spin-coated films of various lamellar-forming phospholipids on solid substrates was previously reported.29–31 Herein, we describe the procedure of spin-coating non-aqueous solutions of SPC and GDO to form thin nonlamellar LC films with controllable properties both on hydrophilic and hydrophobic (dimethyloctylchlorosilanized, DMOCS) silica. The lipids (35/65 wt%/wt% SPC/GDO) were mixed with small fractions of ethanol (EtOH) and propylene glycol (PG) (85/10/5 wt%/wt%/wt% lipid/EtOH/PG) to reduce the viscosity of the formulation and facilitate spreading during the spin-coating procedure. Lipid formulation was added to cover almost the whole substrate surface. Different spinning times were used to prepare films of various thicknesses. After spin-coating the film on the substrate, it was immediately immersed in excess of deionized water, resulting in a negligible concentration of ethanol and propylene glycol in the lipid film. The formation of a lipid LC film was verified by spectroscopic ellipsometry (SE), non-contact mode atomic force microscopy (AFM) and synchrotron small angle X-ray diffraction (SAXD), which provided information on the thickness, topography and LC phase structure of the hydrated lipid film, respectively. A thorough description of the lipid formulation and substrate preparation, the experimental techniques and data analysis is given in the ESI.†
The SE-data recorded in the 620–825 nm wavelength range for a lipid LC film in water at room temperature (∼22 °C) is shown in Fig. 1A. The measured ellipsometric parameters, Δ and Ψ, describe the change in the state of polarization upon reflection of light to a sample surface in terms of phase shift and relative amplitude change (see the ESI† for further details). Single wavelength ellipsometry measurements show, as expected from theoretical modelling, that when the film thickness reaches values at the same order of magnitude as the wavelength of the light, Δ and Ψ oscillate with the film thickness.32 The period of the oscillations in Δ and Ψ depends on the wavelength and SE measurements of thick films thereby giving rise to Δ and Ψ oscillations with the wavelength, where the period and amplitude allow for accurate measurements of the thickness and refractive index.33 The thickness of the hydrated lipid film was obtained by iteratively fitting an optical model to the SE data (Fig. 1A) using a Marquardt least square algorithm. A single optical layer representing the hydrated lipid LC film gave good fit to the experimental data. The model lipid layer was assigned to a refractive index (∼1.465), typical for lipids,34 consistent with the Lorentz oscillator dispersion formula (Fig. SI 1C†) with no absorption of light (k = 0) in the measured spectral range. The fitted thickness of the hydrated lipid film on both hydrophilic and hydrophobic substrates as a function of spinning time is shown in Fig. 1B. Broadening of the interference fringes (not shown) was observed for longer spinning times, as the films got thinner. The thickness of the spin-coated hydrated lipid LC films is highly reproducible and can be controlled with great accuracy, by simply varying the spinning time. The centrifugal force drives the lipid formulation radially outward during spinning. Besides the spinning time and velocity, the viscous flow and solvent evaporation are generally the dominating factors, determining the final film thickness.35 A rapid decrease in film thickness was observed within the first 5–10 minutes of spinning on both substrates (Fig. 1B). On the hydrophobic substrate, the film thickness decreased by 4.4 μm (from 5.6 to 1.2 μm) during the first 10 minutes, while the thickness was only reduced by a further 0.7 μm during the following 30 minutes of spinning. The initial rapid thinning of the film (<∼10 min) is suggested to reflect a period of solvent evaporation, resulting in gradually increasing viscosity and consequently reduced mobility of the lipid formulation. Upon depletion of the solvent (>∼10 min), the viscosity of the film becomes constant. Indeed, the thickness of the film in the region of constant viscosity can be fitted to be inversely proportional to the square root of spinning time (not shown), as expected from models of the spin-coating process.35 The film thickness appears to be independent of substrate properties (Fig. 1B), demonstrating that the formulation bulk properties, compared to formulation/substrate interfacial phenomena, e.g. frictional force, control the final film thickness.
 | ||
| Fig. 1 (A) SE data (Ψ (filled circles) and Δ (open circles)) of a 35/65 wt%/wt% SPC/GDO film in water. The non-aqueous lipid film was spun for 20 s at 8000 rpm and the model fit (lines) revealed a thickness of 5.6 μm. (B) The thickness of lipid LC films on hydrophobic (open symbols) and hydrophilic (filled symbols) substrates in water as a function of spinning time as measured by SE. | ||
A 0.5 μm thick hydrated lipid LC film was prepared on the hydrophobic substrate, while the thinnest film thickness achieved on hydrophilic SiO2 substrates was 2.5 μm. A longer spinning time resulted in breakage (local de-wetting due to autophobicity) of the non-aqueous lipid films.
Due to the nature of lipid LC phases, the hydrated lipid film is expected to contain water. Thus, the fraction of H2O in the film was fitted, using a refractive index of ∼1.33 (Fig. SI 1C†). The fitted volume fraction of water (10–20%) in the lipid films is in agreement with that expected for the fully hydrated bulk I2 (Fd3m) phase of SPC and GDO.20
It should be noted that a slow retraction of the intact non-aqueous lipid film into one or several droplets occurred both on the hydrophilic and hydrophobic substrates after completion of the spinning. Autophobic properties of the non-aqueous lipid formulation, i.e. adsorption of an oriented lipid monolayer at the substrate, which reduces its surface tension below that of the unorganized bulk lipid mixture, cause the surface dewetting.36 Indeed, hydrophobic surface characteristics, i.e. high contact angle of an added H2O droplet, were observed in the dried areas of the initially hydrophilic substrate. Moreover, a thin layer (15–30 Å) was measured by SE in the same regions in air. This is consistent with the formation of a lipid monolayer oriented with the polar lipid heads facing the hydrophilic substrate. The kinetics of the dewetting process of the non-aqueous lipid film is different on hydrophilic and hydrophobic substrates, on which a droplet is formed after 2 minutes and several hours, respectively. In all cases, immediate immersion of the non-aqueous lipid film prevents the dewetting of the surface as the lipids adhere to the substrate upon hydration.
The nanostructure of the thin hydrated lipid LC film (25 °C) on mica was studied with synchrotron SAXD at beamline I911-4, MAX-lab (Fig. 2A and B). The relative positions of the 12 Bragg reflection peaks detected, i.e. √3, √8, √11, √12, √16, √19, √24, √27, √32, √44, √48, and √51, were indexed as the reflections from a face-centred cubic micellar (I2) phase of the Fd3m space group (Fig. 2B). The structure observed for the 35/65 wt%/wt% SPC/GDO bulk phase (Fig. SI 2†) is thus preserved also in the thin film. Furthermore, the 2D detector image (Fig. 2A) shows distinct diffraction intensity spots rather than continuous centrosymmetric circles expected for a polycrystalline bulk phase. This testifies an oligocrystalline structure with a limited number of monocrystalline domains, as expected for LC phases with restricted spatial expansion. This strongly supports that the diffraction pattern does originate from the thin film and not from liquid crystalline structures dispersed in the H2O phase in the SAXD sample cell. The repetitive distance of the I2 phase, the lattice parameter (15.7 nm), is similar to that measured for the bulk LC phase (15.25 nm). The lipid LC films prepared in this study, with thicknesses ranging from 0.5 to 5.6 μm (Fig. 1B), are thus between 30 and 360 repetitions thick.
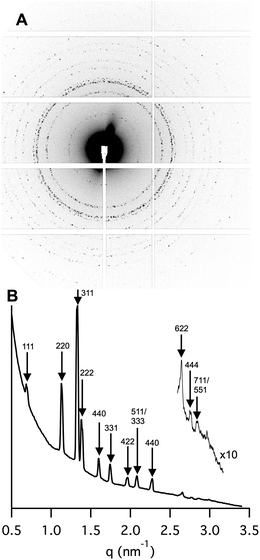 | ||
Fig. 2 (A) The SAXD 2D detector image and (B) the corresponding integrated data of the 35/65 wt%/wt% SPC/GDO film in H2O at 25 °C. High q data are magnified (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) to disclose additional Fd3m reflections. The index corresponding to the face-centred cubic micellar phase is indicated in (B). :10) to disclose additional Fd3m reflections. The index corresponding to the face-centred cubic micellar phase is indicated in (B). | ||
The micro-structured surface topography of the lipid LC film (2.5 μm thick) was mapped by non-contact (tapping) mode AFM (Fig. 3). A similar surface structure was depicted for films on both hydrophobized SiO2 and freshly cleaved mica sheets, confirming the independence of substrate properties, at least for films of this thickness. The surface of the lipid LC film has a grainy appearance at 25 °C. Interestingly, the lateral size (0.5–3 μm) of the grains (Fig. 3C) is in the same order as the size previously reported for monocrystalline domains in similar lipid LC systems.37 Thus, we suggest that the depicted grains are monocrystalline I2 domains, in accordance with the SAXD 2D detector image (Fig. 2A), confirming a limited number of monocrystalline domains in the hydrated lipid LC film at 25 °C.
 | ||
| Fig. 3 Non-contact mode AFM (45 × 45 μm) images of the lipid LC film in H2O on hydrophobic substrates, at (A) 25 and (B) 37 °C. The height profiles along the solid and dashed lines marked in (A) and (B), respectively, are shown in (C). | ||
At physiological temperature, 37 °C, the lipid LC film has a smoother appearance (Fig. 3B), although containing holes with a lateral size of 1–5 μm. SAXD data of the corresponding hydrated bulk lipid LC phase (Fig. SI 2†) show that the highly ordered I2 phase coexists with an increasing fraction of the L2 phase upon heating to 37 and 42 °C, respectively. The AFM images show that the angular domains of the I2 phase fuse and form a smoother surface upon melting of the lipid LC film, which is consistent with the softer and less viscous texture of the disordered L2 phase. The location of the cavities in the film is not understood but could be a consequence of local highly structured I2 regions, still present in the film. Upon re-cooling to 25 °C (Fig. SI 3†), the angular domains are regenerated but are generally larger (laterally: ∼5 μm) than those present prior to heating.
In this communication we report on the controllable formation of thin films of nonlamellar reversed lipid LC phases on solid surfaces. This study also demonstrates that in combination spectroscopic ellipsometry, synchrotron SAXD and AFM can provide comprehensive detailed information on a nonlamellar lipid LC film with potential use as a bioadhesive drug delivery system. They can also be used as versatile model surfaces to study a range of properties of lipid nonlamellar LC phases with relevance to e.g. drug delivery applications. For instance, their adhesion to biological surfaces, interaction with biomolecules and degradation under various simulated physiological conditions can be investigated, with relevance to a range of biomedical applications in mind. A non-aqueous 35/65 wt%/wt% SPC/GDO formulation was spin-coated on solid surfaces to form a thin film. Lipid I2 (Fd3m) films of controllable thicknesses between 0.5 and 5.6 μm were formed upon hydration, as measured by spectroscopic ellipsometry. Such lipid films are only 30–360 repetitive units thick and may thus allow for studies of lipid adhesion to surfaces and its potential effects on the LC phase. This is key knowledge in development of topical drug delivery systems, in which the lipid LC phases are designed to adhere to different biological surfaces, such as oral mucosa in the case of episil®. The potential of using AFM to directly study e.g. microstructural responses of the liquid crystalline phase upon various stimuli was also demonstrated.
Acknowledgements
This work was financed by the Swedish Foundation for Strategic Research (SSF) via framework grant RMA08-0056. Knut and Alice Wallenberg's foundation funded the acquisition of the spectroscopic ellipsometer and the atomic force microscope and the nanometer consortium at Lund University (nmC@LU) helped fund the spin-coater. The authors also thank the Swedish synchrotron X-ray facility MAX-lab for allocating beamtime at the I911-4 beamline and Ana Labrador and Tomás Plivelic for technical support during experiments.Notes and references
- J. M. Lawrence, Chem. Soc. Rev., 1994, 23, 417–424 RSC.
- C. J. Drummond and C. Fong, Curr. Opin. Colloid Interface Sci., 1999, 4, 449–456 CrossRef CAS.
- T. Norling, P. Lading, S. Engstrom, K. Larsson, N. Krog and S. S. Nissen, J. Clin. Periodontol., 1992, 19, 687–692 CrossRef CAS.
- J. Clogston and M. Caffrey, J. Controlled Release, 2005, 107, 97–111 CrossRef CAS.
- M. D. Wyatt and D. Dorschel, Pharm. Technol., 1992, 8, 116–123 Search PubMed.
- F. Tiberg and M. Johnsson, J. Drug Delivery Sci. Technol., 2011, 21, 101–109 CAS.
- J. C. Shah, Y. Sadhale and D. M. Chilukuri, Adv. Drug Delivery Rev., 2001, 47, 229–250 CrossRef CAS.
- F. Tiberg and M. Johnsson, J. Drug Delivery Sci. Technol., 2011, 21, 101–109 CAS.
- J. Barauskas and T. Nylander, in Delivery and controlled release of bioactives in foods and neutraceuticals, ed. N. Garti, Woodhead Publishing Ltd, Cambridge, 2008, p. 107131 Search PubMed.
- K. Thuresson, F. Tiberg, M. Johansson, I. Harwigsson, M. Johnsson and F. Joabsson, Liquid depot formulations, EP1768650, 2005.
- W. Leesajakul, M. Nakano, A. Taniguchi and T. Handa, Colloids Surf., B, 2004, 34, 253–258 CrossRef CAS.
- Episil®, FDA 510(K) number: K101769.
- J. M. Seddon, Biochim. Biophys. Acta, 1990, 1031, 1–69 CrossRef CAS.
- K. Larsson, J. Phys. Chem., 1989, 93, 7304–7314 CrossRef CAS.
- G. Oradd, G. Lindblom, K. Fontell and H. Ljusberg-Wahren, Biophys. J., 1995, 68, 1856–1863 CrossRef CAS.
- J. Barauskas, C. Cervin, M. Jankunec, M. Spandyreva, F. Ribokaite, F. Tiberg and M. Johnsson, Int. J. Pharm., 2010, 391, 284–291 CrossRef CAS.
- J. M. Seddon, Biochemistry, 1990, 29, 7997–8002 CrossRef CAS.
- V. Luzzati, R. Vargas, A. Gulik, P. Mariani, J. M. Seddon and E. Rivas, Biochemistry, 1992, 14, 279–285 CrossRef.
- M. Pouzot, R. Mezzenga, M. Leser, L. Sagalowicz, S. Guillot and O. Glatter, Langmuir, 2007, 23, 9618–9628 CrossRef CAS.
- F. Tiberg, M. johnsson, J. Jankunec and J. Barauskas, Chem. Lett., 2012, 41, 1090–1092 CrossRef CAS.
- J. F. Nagel, Biochim. Biophys. Acta, 2000, 1469, 159–195 CrossRef.
- V. Kiessling, M. K. Domanska, D. Murray, C. Wan and L. K. Tamm, Wiley Encyclopedia of Chemical Biology, John Wiley & Sons, Inc., 2008 Search PubMed.
- R. P. Richter, R. Bérat and A. R. Brisson, Langmuir, 2006, 22, 3497–3505 CrossRef CAS.
- K. B. Blodgett, J. Am. Chem. Soc., 1935, 57, 1007–1022 CrossRef CAS.
- Y. Deng, Z. A. Almsherqi, M. M. Ng and S. D. Kohlwein, Trends Cell Biol., 2012, 20, 371–379 CrossRef.
- P. Vandoolaeghe, R. A. Campbell, A. R. Rennie and T. Nylander, J. Phys. Chem. C, 2009, 113, 4483–4494 CAS.
- C. D. Driever, X. Mulet, A. P. R. Johnston, L. J. Waddington, H. Thissen, F. Caruso and C. J. Drummond, Soft Matter, 2011, 7, 4257–4266 RSC.
- A. G. Emslie, F. T. Bonner and L. G. Peck, J. Appl. Phys., 1958, 29, 858–862 CrossRef CAS.
- U. Mennicke and T. Salditt, Langmuir, 2002, 18, 8172–8177 CrossRef CAS.
- L. Perino-Gallice, G. Fragneto, U. Mennicke, T. Salditt and F. Rieutord, Eur. Phys. J. E: Soft Matter Biol. Phys., 2002, 8, 275–282 CrossRef CAS.
- A. C. Simonsen and L. A. Bagatolli, Langmuir, 2004, 20, 9720–9728 CrossRef CAS.
- R. A. Campbell and K. J. Edler, Soft Matter, 2011, 7, 11125–11132 RSC.
- H. G. Tompkins and W. A. McGahan, Spectroscopic Ellipsometry and Refletctometry, John Wiley & Sons, Inc., New York, 1999 Search PubMed.
- S. F. O'Keefe and O. A. Pike, Fat characterization, in Food Analysis, Food Science Texts Series, ed. S. S. Nielsen, Springer, USA, 2010, ch. 14, pp. 239–260 Search PubMed.
- N. Sahu, B. Parija and S. Panigrahi, Indian J. Phys., 2009, 83, 493–502 CrossRef CAS.
- E. F. Hare and W. A. Zisman, J. Phys. Chem. B, 1955, 59, 335–340 CrossRef CAS.
- J. Barauskas, C. Cervin, F. Tiberg and M. Johnsson, Phys. Chem. Chem. Phys., 2008, 10, 6483–6485 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details about the substrate and sample preparation, the SE and SAXD measurements and AFM imaging. Description of the optical model, used to fit the SE data. SAXD data for the bulk 35/65 wt%/wt% SPC/GDO LC phase at 25, 37 and 42 °C and the calculation of the lattice parameter. See DOI: 10.1039/c3sm51385h |
| This journal is © The Royal Society of Chemistry 2013 |