 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of H-NS on the elongation and compaction of single DNA molecules in a nanospace
Ce Zhang†
a,
Durgarao Guttula†a,
Fan Liua,
Piravi P. Malara,
Siow Yee Nga,
Liang Daib,
Patrick S. Doylebc,
Jeroen A. van Kana and
Johan R. C. van der Maarel*ab
aDepartment of Physics, National University of Singapore, Singapore 117542. E-mail: johanmaarel@gmail.com; Fax: +65 6777612; Tel: +65 65164396
bBioSystems and Micromechanics, Singapore – MIT Alliance for Research and Technology, Singapore
cDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
First published on 14th August 2013
Abstract
The effect of the bacterial heat-stable nucleoid-structuring protein (H-NS) on the conformation of single DNA molecules confined in a nanochannel was investigated with fluorescence microscopy. With increasing concentration of H-NS, the DNA molecules either elongate or contract. The conformational response is related to filamentation of H-NS on DNA through oligomerization and H-NS mediated bridging of distal DNA segments and is controlled by the concentration and ionic composition of the buffer. Confinement in a nanochannel also facilitates compaction of DNA into a condensed form for over-threshold concentrations of H-NS. Divalent ions such as magnesium facilitate but are not required for bridging nor condensation. The time scale of the collapse after exposure to H-NS was determined to be on the order of minutes, which is much shorter than the measured time required for filamentation of around one hour. We found that the effect of H-NS is not only related to its binding properties but also the confinement is of paramount importance. The interplay between confinement, H-NS-mediated attraction, and filamentation controls the conformation and compaction of DNA. This finding might have implications for gene silencing and chromosome organisation, because the cross-sectional dimensions of the channels are comparable to those of the bacterial nucleoid.
1 Introduction
Heat-stable nucleoid-structuring protein (H-NS, 15.6 kDa, pI 7.5) is involved in transcriptional repression (gene silencing) as well as the organisation of the bacterial genome.1 H-NS binds and oligomerizes along double stranded DNA to form a semi-rigid nucleoprotein filament, increases the thermal stability of the duplex, and inhibits transcription.2,3 The histone-like function of H-NS in chromosome organisation is poorly understood. It has been proposed that the compaction into the nucleoid is related to, among others, DNA supercoiling and osmotic stress through macromolecular crowding. Like-charge attraction between distal DNA segments by the binding protein (bridging) is also thought to be important.4 Indeed, it has been shown that H-NS reduces the physical extent of circular plasmids through side-by-side binding of opposing segments.5 Divalent ions such as magnesium and calcium have been reported to play a pivotal role in H-NS mediated bridging of DNA.6 However, H-NS does not behave like a regular condensing multivalent cationic ligand, because it does not compact DNA into a structure with an ordered morphology (condensation) under physiological conditions.7Advances in nanofabrication have made it possible to fabricate quasi one-dimensional channel devices with cross-sectional diameters on the order of tens to hundreds of nanometers. These channels serve as a platform for studying, among others, single DNA molecules.8,9 Furthermore, confinement in a nanospace results in significant modification of certain important biophysical phenomena, such as the knotting probability of circular DNA and the effect of macromolecular crowding.10–13 In particular, it was shown that DNA can be compacted into a condensed form for over-threshold concentrations of dextran or like-charged proteins such as bovine serum albumin and hemoglobin. Here, we report the effect of H-NS in conjunction with confinement inside a nanochannel on the conformation and compaction of DNA. For this purpose, we have done two different, but related series of experiments. In the first series, we focus on equilibrium properties of DNA molecules that have been pre-incubated with H-NS. In the second series, the dynamic, conformational response of DNA molecules immediately following exposure to H-NS is investigated. Super-resolution fluorescence imaging of H-NS in living Escherichia coli cells has shown that it is clustered within the nucleoid with a diameter of a few hundred nanometers.14 This diameter is comparable to the diameters of our channel systems. Accordingly, we surmise that our results for nanochannel confined DNA have implications for gene silencing and chromosome organisation.
For the first series of experiments, T4-DNA (166 kbp) was incubated in buffers of various ionic compositions and various concentrations of H-NS for at least 24 h. Some of the buffers also contained divalent magnesium ions. The DNA molecules were stained with YOYO-1 with an intercalation ratio of 100 base pairs per dye molecule. For such a low level of intercalation, the distortion of the secondary DNA structure is minimal. Furthermore, there is no appreciable effect on the bending rigidity, as inferred from previously reported measurements of the extension of DNA in nanochannels with different concentrations of the dye.15 The pre-incubated molecules were subsequently electrophoresed into a single array of long and rectangular nanochannels with average cross-sectional diameters of 200 or 250 nm. Our chips are made of polydimethylsiloxane (PDMS) cast on a high quality master stamp, obtained by proton beam writing and UV lithography.15–17 The advantage of this technology is that about a hundred replicas can be made with a single stamp, so that a fresh chip can be used for every experiment. Once the pre-incubated molecules are equilibrated after switching off the electric field, their extensions along the direction of the channel (stretch) were measured with fluorescence microscopy. Depending on solution conditions, we observed an elongation or contraction of the DNA molecules with respect to the protein-free state. Furthermore, as in the case of macromolecular crowding, we observed a collapse to a condensed form for over-threshold concentrations of H-NS.
The second series of experiments was done with a recently developed cross-channel device.18 As shown by its layout in Fig. 1, a centrally located grid of rectangular nanochannels is connected to two pairs of loading reservoirs with a set of microchannels. The grid features two arrays of parallel nanochannels in a perpendicular configuration. The average cross-sectional diameters of the channels pertaining to the respective arrays are 225 and 175 nm. DNA immersed in the relevant buffer, but without H-NS, was electrophoresed into the device through the array of wider channels in one direction. Once the molecules were equilibrated inside the channels after switching off the electric field, a buffered solution of H-NS was pipetted into one of the reservoirs of the other set. The protein subsequently diffuses through the intersecting array of channels and uniformly penetrates the array of wider channels. During and following exposure to H-NS, the conformational response of the DNA molecules was monitored with fluorescence microscopy. These experiments were done with a range of H-NS concentrations covering the critical concentration for condensation. Accordingly, the collapse to the condensed form is observed in real time.
 | ||
| Fig. 1 (A) Bright field optical image of the bonded cross-channel array device. The loading reservoirs are connected to the nanochannels by microchannels in SU-8 resin. (B) Optical image of the master stamp featuring the nanochannel grid with connecting microchannels. The channels have a uniform height of 200 nm and widths of 250 and 150 nm in the two perpendicular directions, respectively. The wide and narrow channels are laid on a rectangular grid and separated by 4.0 and 3.75 μm, respectively.18 | ||
Our observations will be analysed in terms of current knowledge of the binding properties of H-NS, in particular filamentation of H-NS on DNA and H-NS mediated like-charge attraction of distal DNA segments. However, it will be shown that, besides binding, confinement is of paramount importance in H-NS mediated control of the conformation and compaction of DNA.
2 Materials and methods
2.1 Isolation and purification of H-NS
K1746 bacteria transformed with pHOP-II were grown on a Luria Broth (LB) plate with ampicillin (50 mg per L). A single colony was taken to grow a starter culture in LB medium containing ampicillin at 310 K for 8 h (OD600 = 0.6). The starter culture was then diluted 1000 times into LB medium containing ampicillin and grown at 310 K for 2 h with vigorous shaking (280 rpm, OD600 = 0.6). Protein expression is induced by addition of isopropylthio-β-galactoside (IPTG) to a concentration of 1 mM. The bacterial cells were harvested by centrifugation at 6000g for 15 min at 277 K. The pellet was suspended in a buffer comprising 20 mM Tris–HCl, pH 7.2, 1 mM EDTA, 5 mM β-mercaptoethanol, 10% glycerol, 100 mg phenylmethylsulfonyl fluoride (PMSF) per L, 2 mM benzamidine, 100 mM NH4Cl, and 1 mg Pefabloc SC per L, lysed with a sonicator (Sonic vibra) at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 joules, and followed by treatment with DNase-1 and RNase-A (Promega) on ice for 30 min. After centrifugation at 15000g for 2.5 h at 277 K, the supernatant was loaded onto a phospho-cellulose (P11, Whatman) column (XK 50/30) equilibrated with 100 mM NH4Cl, 1 mM EDTA, 10% glycerol, 20 mM Tris–HCl, pH 7.2 with an AKTA explorer chromatography system (GE Life Sciences, columns and chromatography media were also purchased from GE). The column was washed with the above-mentioned 100 mM NH4Cl buffer and the protein was eluted with a gradient to 1 M NH4Cl. The elute was subsequently dialysed against 100 mM NaCl, 1 mM EDTA, 10% glycerol, 20 mM Tris–HCl, pH 7.2. The dialysed sample was loaded onto a heparin column equilibrated with the 100 mM NaCl buffer followed by elution in a gradient to 1 M NaCl. Finally, the protein solution was dialysed against 300 mM NaCl and stored at 277 K. At each step, the purity of H-NS was monitored by SDS-PAGE gel chromatography. The secondary structure of H-NS in the prevalent buffers was confirmed with UV circular dichroism measurements.
000 joules, and followed by treatment with DNase-1 and RNase-A (Promega) on ice for 30 min. After centrifugation at 15000g for 2.5 h at 277 K, the supernatant was loaded onto a phospho-cellulose (P11, Whatman) column (XK 50/30) equilibrated with 100 mM NH4Cl, 1 mM EDTA, 10% glycerol, 20 mM Tris–HCl, pH 7.2 with an AKTA explorer chromatography system (GE Life Sciences, columns and chromatography media were also purchased from GE). The column was washed with the above-mentioned 100 mM NH4Cl buffer and the protein was eluted with a gradient to 1 M NH4Cl. The elute was subsequently dialysed against 100 mM NaCl, 1 mM EDTA, 10% glycerol, 20 mM Tris–HCl, pH 7.2. The dialysed sample was loaded onto a heparin column equilibrated with the 100 mM NaCl buffer followed by elution in a gradient to 1 M NaCl. Finally, the protein solution was dialysed against 300 mM NaCl and stored at 277 K. At each step, the purity of H-NS was monitored by SDS-PAGE gel chromatography. The secondary structure of H-NS in the prevalent buffers was confirmed with UV circular dichroism measurements.
2.2 Sample preparation
T4-DNA was purchased from Nippon Gene, Tokyo and used without further purification. The integrity of T4-DNA was verified with pulsed gel electrophoresis. No fragments of ones to tens of kbps were observed. YOYO-1 was purchased from Invitrogen, Carlsbad, CA. T4-DNA was stained with YOYO-1 with an intercalation ratio of 100 base-pairs per dye molecule. No anti-photo-bleaching agent was used, since it might interfere with DNA–protein binding. Samples were prepared by dialysing solutions of DNA against 10 mM Tris–HCl with the relevant concentration of NaCl and/or MgCl2 in micro-dialysers. Solutions of H-NS in the same buffer were also prepared. The Tris–HCl concentration is 10 mM Tris adjusted with HCl to pH 7.5, i.e. 8.1 mM TrisCl and 1.9 mM Tris. The ionic strength of the buffer was calculated with the Davies equation for estimating the activity coefficients of the ions and a dissociation constant pK = 8.08 for Tris. For the measurement of the pre-equilibrated DNA molecules, solutions of H-NS and DNA were subsequently mixed and incubated for 24 h at 277 K. The final DNA concentration is 3 mg per L. Fluorescein 5-isothiocyanate (FITC) was purchased from Sigma-Aldrich. H-NS was labeled with FITC following a standard protocol.19 For the determination of the time required for the protein influx, a solution of 1 μM of FITC labeled H-NS in T-buffer was prepared.2.3 Fabrication of nanofluidic chips
The nanofluidic devices were fabricated by replication in PDMS of patterned master stamps.15,16 The nanochannels were made in HSQ resist (Dow Corning, Midland, MI) using a lithography process with proton beam writing.17 Chips with two different channel layouts were made. For the measurement of the pre-incubated DNA molecules, a single array of nanochannels is connected to two loading reservoirs through a superposing set of microchannels made in SU-8 resin with UV lithography. For the investigation of the response to a change in solution conditions, a cross-channel device was made. In the latter device, there are two intersecting arrays of nanochannels connected to two pairs of loading reservoirs. The heights and widths of the positive channel structures on the stamps were measured with atomic force microscopy (Dimension 3000, Veeco, Woodbury, NY) and scanning electron microscopy, respectively. For the single-array device, two stamps were made featuring nanochannels of length 60 μm and rectangular cross-sections of 150 × 250 and 200 × 300 nm2, respectively. The cross-sections pertaining to the two intersecting arrays of the cross-channel device are 150 × 200 and 200 × 250 nm2. The connecting microchannels have a width and height of 30 and 5 μm, respectively. The stamp was coated with a 5 nm thick Teflon layer to guarantee a perfect release of the replicated PDMS chips.20 The stamps were replicated in PDMS followed by curing with a curing agent (Sylgard, Dow Corning) at 338 K for 24 h. The PDMS replica was sealed with a glass coverslip, after both substrates were plasma oxidised (Harrick, Ossining, NY).2.4 Single-channel array
The pre-incubated and stained DNA molecules dispersed in the relevant solution were loaded into one of the two reservoirs connected to the single array of nanochannels. The DNA molecules were subsequently driven into the channels by electrophoresis. For this purpose, two platinum electrodes were immersed in the reservoirs and connected to an electrophoresis power supply with a relatively low voltage in the range 0.1–10 V (Keithley, Cleveland, OH). Once the DNA molecules were localised inside the nanochannels, the electric field was switched off and the molecules were allowed to relax to their equilibrium state for at least 60 s. The stained DNA molecules were visualised with a Nikon Eclipse Ti inverted fluorescence microscope equipped with a 200 W metal halide lamp, a filter set, and a 100× oil immersion objective. A UV light shutter controlled the exposure time. Images were collected with an electron multiplying charge coupled device (EMCCD) camera (iXon X3, Andor Technology, Belfast, UK) and the extension of the DNA molecules inside the channels was measured with imageJ software (http://rsb.info.nih.gov/ij/). For the intensity threshold, we have used two times the signal to background noise ratio.2.5 Cross-channel array
The protein-free, stained DNA molecules were loaded into the two reservoirs connected to the array of 200 × 250 nm2 nanochannels. To maintain the balance in pressure, T-buffer (without DNA) was loaded into the other two reservoirs connected to the perpendicular, intersecting array of 200 × 150 nm2 channels. The DNA molecules were subsequently driven into the channels by electrophoresis and allowed to relax for at least 60 s as described above. At pre-set times, solutions of H-NS are pipetted into one of the reservoirs of the other set using a microinjector with a minimal pressure of 0.5 kPa (Narishige, Tokyo). The protein is subsequently transported through the microchannel and the intersecting array of 200 × 150 nm2 into the 200 × 250 nm2 nanochannels by the combination of the minimal hydrostatic pressure resulting from the filling of the reservoir and diffusion. During and following exposure to H-NS, the stained DNA molecules were visualised and their extensions were analysed as described above.2.6 Monte Carlo simulation
In the Monte Carlo protocol, the chain is modelled as a string of (N + 1) beads, which are connected by N inextensible bonds of length lb.21,22 Furthermore, the model consists of bond bending energy, hard-sphere repulsion between beads, and hard-wall repulsion between the beads and the wall. If the centre of a bead is beyond the channel wall, the potential becomes infinitely large and the configuration is rejected. The effective channel diameter is hence the real diameter minus the diameter of the bead. The diameter of the bead was set equal to the bond length lb, which is equivalent to the chain width w. We have done two series of simulations. In the first series, the bending rigidity is set to reproduce a persistence length P = 50 nm. The contour length of the chain with widths w = 5, 7.5, or 10 nm was fixed at L = 8 μm. The second series was done for a range in persistence length P = 50–160 nm, but with a single width w = 10 nm and contour length L = 16 μm. In each Monte Carlo cycle, we carried out one crankshaft and one reptation move. The simulation started from a random conformation and reached equilibrium after 107 cycles. In the production run, we generated 1010 cycles and recorded the conformation every other 105 cycles. For each conformation, we calculated the extension as the maximum span of the molecule along the channel axis. Finally, the extension was averaged over the ensemble of 105 conformations. We have verified that effects of finite contour length on the relative stretch are unimportant for the relevant range of channel diameters.3 Results
3.1 Stretch of pre-incubated DNA
In the first series of experiments, T4-DNA molecules were incubated with the relevant buffer for at least 24 h before they were brought into the channels of the single-array device. Montages of images of single DNA molecules confined in rectangular channels with a cross-section of 200 × 300 nm2 are shown in Fig. 2. The images refer to well-equilibrated conformations. After the electric field has been switched off, the molecules relax to their equilibrium state within 60 s. We have verified that there is no further change in the extension of the molecules for more than 3 h. Furthermore, we observed no difference in extension between molecules inserted by electrophoresis or pressure. The equilibrated stretch in the longitudinal direction of the channel depends on the buffer conditions. In a buffer with a moderate ionic strength of around 11 mM (T-buffer with 3 mM NaCl, T-buffer is 8.1 mM TrisCl and 1.9 mM Tris, pH 7.5), the DNA molecules elongate with increasing concentration of H-NS. In the case of a buffer with a higher ionic strength of about 38 mM, the molecules contract with respect to the protein-free situation. In the presence of sub-millimolar concentrations of magnesium ions, the H-NS induced contraction is minimal, if not negligible. For over-threshold concentrations of H-NS, condensation of the DNA molecules into a compact form is observed. Condensed DNA is visible as a bright fluorescence spot and can easily be discerned from the extended form. In channels with a smaller cross-section of 150 × 250 nm2, we observed the same qualitative behaviour. There are quantitative differences however in the values of the stretch and critical concentration of H-NS for condensation. Note that differences in width and brightness between molecules of similar extension are related to effects of photo-bleaching and have no physical meaning. | ||
| Fig. 2 (A) Montage of fluorescence images of T4-DNA in T-buffer with 3 mM NaCl and inside 200 × 300 nm2 channels. T-buffer is 8.1 mM TrisCl and 1.9 mM Tris, pH 7.5. The H-NS concentration is 0, 0.3, and 0.6 μM from left to right. The scale bar denotes 3 μm. (B) As in panel (A), but in T-buffer with 30 mM NaCl. The H-NS concentrations are 0 and 0.3 μM from left to right. (C) As in panel (A), but in T-buffer with 3 mM NaCl and 0.2 mM MgCl2. The H-NS concentrations are 0 and 0.1 μM from left to right. (D) Distribution in extension of a population of 50 molecules in T-buffer with 3 mM NaCl and 0.3 μM H-NS, inside 200 × 300 nm2 channels, A Gaussian fit gives R‖ = 10 ± 2 μm. | ||
We have measured the extension of the DNA molecules in channels with two different cross-sections: 200 × 300 and 150 × 250 nm2. For each experimental condition, that is buffer composition, channel diameter, and H-NS concentration, we have used a fresh PDMS replica and measured around 50 molecules. The distribution in extension is close to Gaussian.23 An example of such a distribution is also shown in Fig. 2. Fragmented DNAs can easily be discerned, because their extensions clearly fall below the values pertaining to the intact molecules. For the cut-off, we have used the mean value minus two times the standard deviation. Resolution broadening can be neglected, because the optical resolution is one order of magnitude smaller than the variance. The mean relative extension R‖/L, that is the mean extension divided by the YOYO-1 corrected contour length of 57 μm, is shown in Fig. 3 as a function of the H-NS concentration. An enhanced stretch is observed if the molecules are bathed in a buffer of moderate ionic strength without divalent ions (T-buffer with 3 mM NaCl). In the case of a buffer of higher ionic strength (T-buffer with 30 mM NaCl), different behaviour is observed. Now the molecules contract with increasing concentration of H-NS. The stretch is constant or slightly decreases with increasing H-NS concentration in the presence of 0.2–0.8 mM MgCl2. Note that for sub-threshold concentrations of H-NS the relative extensions are in the range of 0.05–0.4, which implies that the DNA molecules remain coiled. Furthermore, related to the stronger confinement, the stretch is more pronounced in the 150 × 250 nm2 channel system.
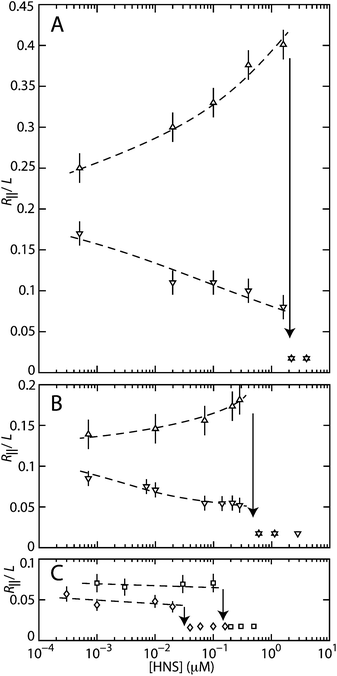 | ||
| Fig. 3 (A) Relative extension R‖/L of T4-DNA in T-buffer with 3 (△) or 30 (▽) mM NaCl versus the concentration of H-NS. The molecules are inside 150 × 250 nm2 channels. (B) As in panel (A), but in 200 × 300 nm2 channels. (C) As in panel (A), but in T-buffer with 3 mM NaCl and 0.2 (□) or 0.8 (◇) mM MgCl2, inside 200 × 300 nm2 channels. The dashed curves are drawn as an aid to the eye and the arrows denote the condensation thresholds. | ||
For over-threshold concentrations of H-NS, the DNA molecules compact into a condensed form. This is facilitated by the confinement in the nanochannel, because we did not observe condensation in the feeding microchannels and/or the reservoirs of the chip. To the best of our knowledge, condensation of DNA by H-NS has not been reported before. In a buffer composed of monovalent salts, the critical concentrations of H-NS for condensation are 0.5 ± 0.2 and 1.9 ± 0.3 μM H-NS for the 200 × 300 and 150 × 250 nm2 channel systems, respectively. The threshold shifts hence to a higher value with decreasing channel cross-sectional diameter. There is however no significant effect of the concentration of monovalent salts. On the other hand, the critical concentration for condensation is shifted downwards from 0.5 to 0.03–0.1 μM of H-NS in the presence of sub-millimolar concentrations of magnesium ions.
3.2 Time-dependent conformational response
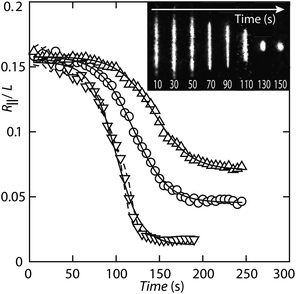
The above described results refer to DNA molecules pre-incubated with H-NS for more than 24 h before they were brought into the nanochannels of the single-array device. In order to obtain more insight into the various mechanisms at hand, we have done a second series of experiments with the recently developed cross-channel device.18 With the latter device, the conformational response of the DNA molecules to a change in environmental solution conditions can be investigated in situ. To avoid complications related to contraction prior to the collapse, we have used a buffer of moderate ionic strength and without divalent ions (T-buffer with 3 mM NaCl). In such a buffer, the pre-incubated DNA molecules are either elongated or condensed, depending on the concentration of H-NS. To cover the elongation and condensation phenomena, we have done a series of experiments with a range of concentrations of H-NS across the critical concentration for condensation (0.8–1.6 μM).Protein-free DNA molecules were electrophoresed into the array of wider 200 × 250 nm2 channels of the cross-channel device. Once the electric field is removed, the molecules equilibrate and remain stationary. The initial stretch is around 9 μm. The H-NS containing buffer was pipetted into one of the reservoirs of the other set and diffused through the intersecting array of narrower 150 × 200 nm2 channels into the wider channels. Throughout the exposure to H-NS, the DNA molecules are imaged with fluorescence microscopy and were observed to remain inside the array of wider channels. The result pertaining to elongation with 0.8 μM H-NS is shown in Fig. 4. Condensation following exposure to over-threshold concentrations of H-NS (1.0, 1.2, and 1.6 μM) is illustrated in Fig. 5. The threshold concentration for condensation inside the 200 × 250 nm2 channels is 0.9 μM H-NS. This value falls between the thresholds for pre-incubated DNA in slightly wider and narrower channels. We observed that the molecules remain mobile and that there is no H-NS-mediated sticking of DNA to the channel walls.
 | ||
| Fig. 4 Elongation of T4-DNA following exposure to 0.8 μM H-NS in T-buffer and 3 mM NaCl. The molecules are inside 200 × 250 nm2 channels and, initially, in T-buffer with 3 mM NaCl. The dashed curves pertain to the elongation of three different molecules and the symbols represent the average. The solid curve represents a sigmoidal fit. The inset shows a time-lapse series of fluorescence images pertaining to a single molecule. | ||
 | ||
| Fig. 5 Condensation of T4-DNA following exposure to 1.0 (△), 1.2 (○), and 1.6 (▽) μM H-NS in T-buffer with 3 mM NaCl. The molecules are inside 200 × 250 nm2 channels and, initially, in T-buffer with 3 mM NaCl. The symbols represent averages pertaining to two different molecules for each buffer condition. Individual trajectories are denoted by the dashed curves (1.6 μM H-NS only). The solid curves represent sigmoidal fits. The inset shows a time-lapse series of fluorescence images showing the compaction of a single molecule into the condensed form following exposure to 1.6 μM H-NS. | ||
In order to gauge the conformational response, it is necessary to determine the time required for the influx of protein. For this purpose, 1 μM FITC-labelled H-NS was pipetted into one of the loading reservoirs connected to the array of narrow nanochannels. With fluorescence microscopy, it was monitored that the protein is transported through the microchannel and reaches the entrance of the nanochannels in about 30 s. Subsequently, the protein diffused through the narrow channels into the wide channels.18 Fig. 6 shows the time evolution of the integrated fluorescence intensity across the array of narrow channels. The buffer is progressively and uniformly replaced within about 3 s from the moment H-NS enters the array of nanochannels. Eventually, the protein exits through the nanochannels into the microchannels connected to the other reservoirs. We have verified that, due to the continuous influx of H-NS, there is no appreciable drop in the integrated fluorescence intensity and, hence, the protein concentration for longer times. Furthermore, we did not observe adhesion of H-NS to the surface of PDMS.
 | ||
| Fig. 6 (A) Fluorescence intensity profile across the array of 200 × 150 nm2 channels imaged during the exchange of FITC-labelled H-NS. The channels are separated by 3.75 μm. (B) Integrated fluorescence intensity as a function of elapsed time. Time zero is defined as the moment when the protein enters the nanochannels and the fluorescence intensity starts to increase. | ||
As can be seen in the time-lapse series of fluorescence images and relative extensions in Fig. 4, for a sub-threshold concentration of H-NS the DNA molecules elongate in a sigmoidal fashion. We observed some variation in elongation pertaining to different molecules, but, overall, the rate and lag-time are 0.06 ± 0.01 min−1 and 42 ± 2 min, respectively. The molecules reach the final stretch in around 90 min. The lag-time is much longer than the time required for the influx of protein (<3 s). All molecules reach a final extension of around 15 μm. With an extension prior to exposure to H-NS of around 9 μm, the stretch has increased by about 70%. The final extension falls between the extensions just prior to the collapse of pre-incubated DNA in slightly wider and narrower channels.
A time lapse series of fluorescence images captured after exposure to over-threshold concentrations of H-NS, as well as the corresponding relative extensions, are shown in Fig. 5. We have averaged the results obtained from two different DNA molecules for each buffer condition, because individual trajectories coincide within experimental error. The extensions decrease in a sigmoidal fashion and, eventually, level off at a level pertaining to the condensed form. The relative decrease in extension ΔR‖/L, decay rate R, and lag-time τlag resulting from a fit of a sigmoid to the data are collected in Table 1. With increasing concentration of H-NS, the rate and lag-time increases and decreases, respectively. Besides compaction time, the final extension of the condensed molecules inside the nanochannels depends on the concentration of H-NS. For 1.0, 1.2, and 1.6 μM H-NS, the final extension is 4.0, 2.6, and 0.9 μm, respectively. We have verified that there is no further decrease in extension for another 2 h.
| ΔR‖/L | R (s−1) | τlag (s) | |
|---|---|---|---|
| 1.0 μM H-NS | 0.087 ± 0.002 | 0.048 ± 0.002 | 144 ± 2 |
| 1.2 μM H-NS | 0.112 ± 0.002 | 0.052 ± 0.002 | 120 ± 2 |
| 1.6 μM H-NS | 0.141 ± 0.004 | 0.069 ± 0.004 | 100 ± 2 |
3.3 Summary of observations
For sub-threshold concentrations of H-NS, the molecules are either elongated or contracted with respect to the protein-free state. Elongation occurs in a monovalent buffer of moderate ionic strength. The final stretch is reached in about 90 min after exposure to H-NS. At higher ionic strength, the molecules contract. In the presence of sub-millimolar concentrations of magnesium ions, the H-NS induced contraction is minimal, if not negligible. DNA compacts into a condensed form for over-threshold concentrations of H-NS. The critical concentration depends on the channel diameter (lower for wider channels), divalent salt (lower for sub-millimolar magnesium), but does not significantly depend on the concentration of monovalent salts. The time required for condensation after an exchange of buffer with H-NS is around a minute. Furthermore, the compaction time and the final extension of the condensates inside the channels depend on the concentration of H-NS, with shorter times and smaller extensions for higher concentrations of H-NS. We did not observe condensation of DNA with H-NS in the bulk phase and/or the microchannels of the chip under the prevalent solution conditions.4 Discussion
4.1 Elongation by filamentation
H-NS is known to bind on DNA and oligomerizes along the contour to form a nucleoprotein filament.3 The filamentation takes typically a few hours.6 Furthermore, it has been shown that the persistence length P increases from the nominal value of 50 nm for bare DNA to around 140 nm in the presence of 4.0 μM H-NS and 50 mM KCl.2 With a DNA concentration of 3 mg L−1, an H-NS concentration of 4.5 μM corresponds with one H-NS dimer per base pair. Accordingly, with a range of four decades in the H-NS dimer to base pair ratio, we cover the situation with sparsely bound H-NS on DNA for the lowest concentrations of H-NS to a fully coated filament at H-NS concentrations exceeding around 1.0 μM. The observed elongation in monovalent buffer of moderate ionic strength over a time span of 90 min is almost certainly due to filamentation with a concomitant increase in bending rigidity (stiffening). In order to verify this conjecture, we need to relate the stretch of the filament to its persistence length P. For this purpose, we have done Monte Carlo computer simulation of a wormlike chain confined in nanochannels of various cross-sectional diameters.21,22For a self-avoiding, wormlike chain with persistence length P and width w in a wide channel with diameter D ≫ P, Daoud and de Gennes' blob model predicts a scaling law R‖/L ∝ D−2/3P1/3w1/3.24–26 In particular, the scaling exponent for both P and w is predicted to be 1/3. The Monte Carlo simulation results for R‖/L as a function of P for fixed w = 10 nm as well as R‖/L as a function of w for fixed P = 50 nm are shown in Fig. 7. To a good approximation, the extension follows a power law in P and w, that is R‖/L ∝ Pαwβ. The fitted values of the exponent α depend on the value of the tube diameter D. Only for very wide channels with D = 400 nm, the simulation result for α approaches the prediction of blob theory. On the other hand, the simulation results for β are always close to the value given by the blob model, irrespective of the channel diameter.
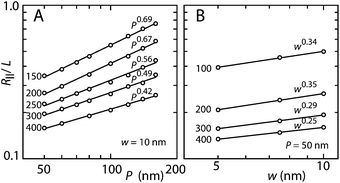 | ||
| Fig. 7 (A) Monte Carlo results of the relative extension R‖/L versus persistence length P for a chain with cross-sectional diameter w = 10 nm. (B) As in panel (A), but for the relative extension versus w for a chain with P = 50 nm. The channel diameters and fitted scaling exponents are indicated. | ||
The width of the filament is expected to be around 10 nm, as estimated from the diameter of the duplex (2 nm) and the diameter of H-NS (3.5 nm). In the absence of more detailed knowledge about the structure of the filament, we ignore a possible dependence of w on the concentration of H-NS. For the interpretation of the elongated stretch in terms of an increase in P, we accordingly use the Monte Carlo results obtained for w = 10 nm. The scaling exponent for P takes the values α = 0.56 and 0.67 in channels with cross-sectional diameters of 250 and 200 nm, respectively. The increase in P with increasing H-NS concentration can then be obtained from P/P∘ = (R∥/R∘∥)1/α, with P∘ and R∘∥ being the persistence length and stretch for the protein-free state in otherwise the same solution conditions, respectively. The results are shown in Fig. 8. With increasing concentration of H-NS, P increases from the value pertaining to bare DNA (60 nm in 10 mM salt26,27) to about 130 nm just prior to the collapse to the condensed form. This increase in P is in good agreement with the one reported in the literature obtained in 50 mM KCl.2 Furthermore, the agreement of P obtained for DNA in channels of two different cross-sections confirms the Monte Carlo results for the scaling exponent α and the cancellation of unknown, channel width dependent pre-factors in the relevant ratio of the extensions.
 | ||
| Fig. 8 Relative persistence length P/P∘ of T4-DNA in T-buffer with 3 mM NaCl and confined in 150 × 250 (▲) and 200 × 300 (△) nm2 channels versus the concentration of H-NS. The dashed line is drawn as an aid to the eye. | ||
Elongation of nano-confined DNA by crowding with dextran was reported before.11–13 This effect was interpreted in terms of depletion of DNA segments and volume occupancy by neutral nanoparticles in the interfacial region next to the channel wall. Depletion can also play a role in the elongation of the nucleoprotein filament. However, such a role is minor at best, because the elongation of DNA induced by H-NS is more pronounced despite the fact that the concentrations of H-NS are an order of magnitude lower than those of dextran. Furthermore, unlike the situation for dextran, the molecules contract in a buffer of higher ionic strength.
4.2 Contraction by bridging
Another aspect is the disappearance of the elongation in the presence of magnesium ions and the contraction in a monovalent buffer of higher ionic strength. An increase in ionic strength reduces the binding efficiency of H-NS.28,29 However, in the present range of salt concentrations (below 50 mM) the concomitant effect on the bending rigidity of the filament is moderate.2 Furthermore, an explanation of a contraction with respect to the protein-free state in terms of a decrease in bending rigidity requires a value of P less than the one pertaining to bare DNA. Although this can be envisaged by, for instance, sharp bends or kinks in the duplex, a more plausible explanation is H-NS mediated side-by-side binding of distal segments of the DNA molecule (bridging). In atomic force microscopy and single-molecule manipulation studies, bridging was observed to be induced by divalent ions such as magnesium and calcium.5,6 Here, we show that for DNA in a nanochannel the switch is merely controlled by screened electrostatics through a variation in the concentration of monovalent salt. The enhanced sensitivity of the conformation of the filament to the concentration and ionic composition of the buffer is plausibly related to segment orientation order induced by confinement (see below).Nano-confined DNA was previously observed to contract in the presence of bovine serum albumin or hemoglobin, irrespective of the ionic strength.13 The latter contraction was explained by depletion of like-charged protein in the interior of the DNA coil and the concomitant osmotic pressure gradient. Since H-NS is net electroneutral at pH 7.5,30 such a mechanism does not explain the switch from elongation to contraction with a change in the ionic composition and/or concentration of the buffer.
4.3 Condensation
A unique feature of the nano-confinement is the compaction of DNA into a condensed form for over-threshold concentrations of H-NS. Condensation of DNA inside a nanochannel by crowding with dextran as well as like-charged proteins has been reported before.11,13 A fundamental difference is the critical concentration of the crowder or like-charged protein in the range of tens to hundreds of micromolars. In the case of H-NS in monovalent buffer, the critical concentration is at least one order of magnitude less and around one micromolar. This concentration corresponds to that of a fully coated filament. The threshold concentration of the filament (pre-incubated DNA) is however about the same as the one observed for bare DNA following exposure to H-NS. Furthermore, the time scale of filamentation (hour) is much longer than the one for condensation (minutes). Accordingly, condensation is not related to filamentation per se, but to H-NS mediated attraction of like-charged, distal DNA segments. This is also supported by the downward shift in the threshold concentration in the presence of magnesium ions, which are known to promote the formation of H-NS bridges.5,6 In this respect, H-NS acts as a condensing agent such as protamine, despite its net neutral charge. Indeed, the threshold concentration and final extension of the condensates for H-NS are similar to those obtained for protamine in the same cross-channel device.18 Protamine induced condensation occurs however markedly faster on a time scale of a few seconds. Another important difference is that H-NS only facilitates condensation once the molecule is confined in a nanospace, whereas protamine also condenses DNA in the bulk phase.31An effective attractive interaction by bridging ligands requires the juxtaposition of two almost parallel DNA segments.32 Once the segments are skewed, the contact area is reduced and the attraction disappears. A plausible mechanism for the nanochannel-facilitated condensation is the increase in contact pairs of (almost) parallel-aligned and juxtaposed segments due to orientation order imposed by the channel walls. The critical concentration for condensation hence depends on two factors: the orientation order and probability of a contact. With decreasing channel diameter, the orientation order increases. Concurrently, the contact probability decreases, because the correlation length of the volume interaction is on the order of the channel diameter. In the blob model, the contact probability is proportional to the number of segments per blob, that is ∝D5/3 with diameter D.24 The increase in critical concentration with decreasing channel diameter can hence be explained by a decrease in contact probability despite the increase in orientation order.
The decrease in extension and eventual condensation following exposure to over-threshold concentrations of H-NS is sigmoidal (Fig. 5). In a coarse grained simulation model, it was seen that DNA compaction is controlled by competing factors.33 These factors include several binding modes of H-NS to DNA and confinement to a planar interface. In particular, it was observed that additional bridges are preferentially formed at sites close to a first bridge, provided that other H-NS dimers are available. The first bridge accordingly acts as a seed to propagate compaction. Such a mechanism in which a dimer D is converted into a bridge B can be represented by an auto-catalysed reaction
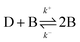 | (1) |
 | (2) |
Condensed DNA has usually an ordered morphology, in which the segments are arranged in a hexagonal fashion.7,34 The structural arrangement of compacted molecules inside nanofluidic channels is probably also hexagonal. This could, however, not be confirmed because of difficulties associated with molecular imaging of enclosed molecules.
5 Conclusions
Our observations on nanochannel-confined DNA agree with the two main features of H-NS, that is filamentation of H-NS on DNA and H-NS mediated bridging of distal DNA segments. Filamentation with a concomitant increase in bending rigidity occurs over a time span of about an hour, as determined from the elongation of DNA along the direction of the channel in a buffer of moderate ionic strength. Elongation is suppressed and the DNA molecule may even contract once bridging comes into play. For DNA in a nanospace, bridging is not only induced by divalent ions such as magnesium, but also by screened electrostatics through the concentration of monovalent salts. Another unique feature of nano-confinement, in conjunction with H-NS mediated bridging, is the collapse to a condensed form for over-threshold concentrations of H-NS. The collapse occurs within a few minutes following exposure to H-NS, which shows that it is not related to filamentation per se. As for sub-threshold suppression of elongation and contraction, divalent ions facilitate but are not required for DNA condensation. The enhanced sensitivity of the conformation to H-NS mediated attraction is plausibly related to DNA segment orientation order induced by confinement inside the channels with cross-sectional diameters on the order of a few times the DNA persistence length. The architectural role of H-NS is hence not only related to its binding modes but also DNA conformation as affected by one or two-dimensional confinement is of paramount importance.33 The interplay between confinement, H-NS-mediated bridging, and filamentation controls the conformation and compaction of DNA. Furthermore, since the typical dimensions of the bacterial nucleoid are comparable to those of our channel systems, these specific effects are likely to play a role in H-NS mediated gene silencing and chromosome organisation.1,3,14Acknowledgements
Nora Goosen is thanked for her gift of the H-NS expression vector. This work is supported by the Singapore – MIT Alliance for Research and Technology (SMART) and National Science Foundation (NSF) grant [CBET-0852235] and the Singapore Ministry of Education grants [R-144-000-270-112 and R-144-000-312-112].References
- D. F. Browning, D. C. Grainger and S. J. W. Busby, Curr. Opin. Microbiol., 2010, 13, 773–780 CrossRef CAS PubMed.
- R. Amit, A. B. Oppenheim and J. Stavans, Biophys. J., 2003, 84, 2467–2473 CrossRef CAS.
- E. Bouffartigues, M. Buckle, C. Badaut, A. Travers and S. Rimsky, Nat. Struct. Mol. Biol., 2007, 14, 441–448 CAS.
- C. L. Woldringh and N. Nanninga, J. Struct. Biol., 2006, 156, 273–283 CrossRef CAS PubMed.
- R. T. Dame, C. Wyman and N. Goosen, Nucleic Acids Res., 2000, 28, 3504–3510 CrossRef CAS PubMed.
- Y. Liu, H. Chen, L. J. Kenney and J. Yan, Genes Dev., 2010, 24, 339–344 CrossRef CAS PubMed.
- V. A. Bloomfield, Curr. Opin. Struct. Biol., 1996, 6, 334–341 CrossRef CAS.
- S. L. Levy and H. G. Craighead, Chem. Soc. Rev., 2010, 39, 1133–1152 RSC.
- W. Reisner, J. N. Pedersen and R. H. Austin, Rep. Prog. Phys., 2012, 75, 106601 CrossRef PubMed.
- L. Dai, J. R. C. van der Maarel and P. S. Doyle, ACS Macro Lett., 2012, 1, 732–736 CrossRef CAS.
- C. Zhang, P. G. Shao, J. A. van Kan and J. R. C. van der Maarel, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16651–16656 CrossRef CAS PubMed.
- J. J. Jones, J. R. C. van der Maarel and P. S. Doyle, Nano Lett., 2011, 11, 5047–5053 CrossRef CAS PubMed.
- C. Zhang, Z. Y. Gong, D. Guttula, P. P. Malar, J. A. van Kan, P. S. Doyle and J. R. C. van der Maarel, J. Phys. Chem. B, 2012, 116, 3031–3036 CrossRef CAS PubMed.
- W. Wang, G. W. Li, C. Chen, X. S. Xie and X. Zhuang, Science, 2011, 333, 1445–1449 CrossRef CAS PubMed.
- C. Zhang, F. Zhang, J. A. van Kan and J. R. C. van der Maarel, J. Chem. Phys., 2008, 128, 225109 CrossRef PubMed.
- J. A. van Kan, C. Zhang, P. P. Malar and J. R. C. van der Maarel, Biomicrofluidics, 2012, 6, 036502 CrossRef PubMed.
- J. A. van Kan, A. A. Bettiol and F. Watt, Nano Lett., 2006, 6, 579–582 CrossRef CAS PubMed.
- C. Zhang, K. Jiang, F. Liu, P. S. Doyle, J. A. van Kan and J. R. C. van der Maarel, Lab Chip, 2013, 13, 2821–2826 RSC.
- Y. Egawa, R. Hayashida, T. Seki and J. Anzai, Talanta, 2008, 15, 736–741 CrossRef PubMed.
- J. A. van Kan, P. G. Shao, Y. H. Wang and P. Malar, Microsyst. Technol., 2011, 17, 1519–1527 CrossRef.
- L. Dai, J. J. Jones, J. R. C. van der Maarel and P. S. Doyle, Soft Matter, 2012, 8, 2972–2982 RSC.
- L. Dai, S. Y. Ng, P. S. Doyle and J. R. C. van der Maarel, ACS Macro Lett., 2012, 1, 1046–1050 CrossRef CAS.
- J. O. Tegenfeldt, C. Prinz, H. Cao, S. Chou, W. W. Reisner, R. Riehn, Y. M. Wang, E. C. Cox, J. C. Sturm, P. Silberzan and R. H. Austin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10979–10983 CrossRef CAS PubMed.
- M. Daoud and P. G. de Gennes, J. Phys., 1977, 38, 85–93 CAS.
- P. G. de Gennes, Scaling Concepts in Polymer Physics, Cornell Univ. Press, Ithaca, NY, 1979 Search PubMed.
- J. R. C. van der Maarel, Introduction to Biopolymer Physics, World Scientific, Singapore, 2008 Search PubMed.
- C. G. Baumann, S. B. Smith, V. A. Bloomfield and C. Bustamante, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6185–6190 CrossRef CAS.
- A. E. Tupper, T. A. Owen-Hughes, D. W. Ussery, D. S. Santos, D. J. Ferguson, J. M. Sidebotham, J. C. Hinton and C. F. Higgins, EMBO J., 1994, 13, 258–268 CAS.
- S. Stella, M. Falconi, M. Lammi, C. O. Gualerzi and C. L. Pon, J. Mol. Biol., 2006, 355, 169–174 CrossRef CAS PubMed.
- A. Spassky, S. Rimsky, H. Garreau and H. Buc, Nucleic Acids Res., 1984, 12, 5321–5340 CrossRef CAS PubMed.
- N. Makita, Y. Yoshikawa, Y. Takenaka, T. Sakaue, M. Suzuki, C. Watanabe, T. Kanai, T. Kanbe, T. Imanaka and K. Yoshikawa, J. Phys. Chem. B, 2011, 115, 4453–4459 CrossRef CAS PubMed.
- L. Dai, Y. Mu, L. Nordenskiöld and J. R. C. van der Maarel, Phys. Rev. Lett., 2008, 100, 118301 CrossRef.
- M. Joyeux and J. Vreede, Biophys. J., 2013, 104, 1615–1622 CrossRef CAS PubMed.
- N. V. Hud and I. D. Vilfan, Annu. Rev. Biophys. Biomol. Struct., 2005, 34, 295–318 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |