 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The dendrimer impact on vesicles can be tuned based on the lipid bilayer charge and the presence of albumin†
Francesca
Ruggeri
a,
Anna
Åkesson
a,
Pierre-Yves
Chapuis
a,
Catherine Anna
Skrzynski Nielsen
b,
Marco P.
Monopoli
c,
Kenneth A.
Dawson
c,
Thomas Günther
Pomorski
b and
Marité
Cárdenas
*a
aInstitute of Chemistry and Nano-Science Center, University of Copenhagen, Universitetparken 5, DK 2100, Copenhagen, Denmark. E-mail: cardenas@nano.ku.dk
bCenter for Membrane Pumps in Cells and Disease – PUMPKIN, Department of Plant and Environmental Sciences, Faculty of Science, University of Copenhagen, Thorvaldsenvej 40, DK-1871 Frederiksberg C, Denmark
cCentre for BioNano Interactions, School of Chemistry and Chemical Biology, UCD Conway Institute for Biomolecular and Biomedical Research, University College Dublin, Belfield, Dublin 4, Ireland
First published on 25th July 2013
Abstract
PAMAM (polyamidoamine) dendrimers are promising polymers in biomedical applications that can interact with both the lipid bilayer and proteins. Here we employed giant unilamellar vesicles (GUVs) of two different charge densities to study the effect of albumin, one of the major proteins in blood plasma, on the interactions between PAMAM dendrimers and lipid membranes. The results show that albumin exacerbates the effect of dendrimers on the destabilization of the vesicles in terms of leakage, aggregation and collapse in particular for negatively charged vesicles while neutrally charged membranes are not affected. We conclude that the higher affinity of both albumin and PAMAM G6 towards negatively charged membranes explains their synergistic behavior in this case. In the case of neutral vesicles, the affinity between PAMAM G6 and albumin is stronger than that between PAMAM G6 (or albumin) and neutral vesicles, and thus no synergism is observed for the mixture during the interaction with neutral membranes.
Introduction
Nanoscaled objects have great potential in different biomedical applications, ranging from drug1–6 and gene delivery7 to magnetic resonance imaging.8 In particular, polyamidoamine (PAMAM) dendrimers have great potential in this field since they are nano-sized polymers with unique properties in terms of monodispersity and ability to tune their overall dimension and surface charge. The capability of PAMAM dendrimers to translocate and deliver a drug/gene has been widely studied using in vitro models based on both lipid membranes and living cells. For example in studies on model membranes, PAMAM dendrimers were shown to bind, due to electrostatic forces and entropic contributions, to zwitterionic and oppositely charged lipid membranes. The net effect of dendrimers binding to lipid membranes has been quite under dispute. Several different mechanisms of interactions have been proposed based on observations of dendrimers opening holes in supported lipid membranes,9–13 among other experimental and computational evidence.14–17 This was interpreted either as dendrimer translocation across lipid membranes due to diffusion across the membrane18 or due to the formation of lipid–dendrimer complexes called “dendrisomes”.19 We have, on the other hand, found no evidence for dendrisome formation or for any type of translocation across lipid bilayers for PAMAM G6 dendrimers, regardless of the charge of the lipid membrane or ionic strength. We have found though that dendrimers adsorb on the surface of lipid vesicles,20,21 and that their adsorption is enhanced in the presence of charged lipids.21,22 At certain dendrimers to lipid ratios, the vesicles collapse21 and a phase separation process takes place in the bulk with the formation of a lamellar type liquid crystalline phase.20 Studies on living cells demonstrated that dendrimers attach to plasma membranes and induce large morphological changes,23,24 and also that they can enter cells by endocytosis.25,26On the other hand, PAMAM dendrimers form complexes with soluble proteins,27–31 including those present in blood plasma.32,33 Thus, dendrimers potentially interact with many components in a living organism besides the cell membrane. They can for instance bind complement proteins,32 thereby causing activation of the immune complement system,34 and induce aggregation of fibrinogen in blood.33Protein binding is indeed a process generalized for all types of nano-scaled particles.35 Consequently, the properties of the proteins rather than the pristine nanoparticle may determine their interaction with cells.
The interaction of G6 PAMAM dendrimers with serum albumin as well as with lipid membranes has been thoroughly studied at the molecular level. In contrast, the dendrimer interaction with lipid bilayers in the presence of soluble plasma proteins including albumin has been largely unexplored to date even though the protein binding might alter the physical–chemical properties of dendrimers. In the present study, we employed, as simple model cell membranes, giant unilamellar vesicles (GUVs) composed of neutral lipids (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, POPC) and mixtures of neutral (POPC) and negatively charged lipids (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-1′-rac-glycerol, POPG, or 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine, POPS) to study the effect of bovine serum albumin (BSA) on the interaction between PAMAM dendrimers and lipid vesicles. Our studies indicate that the impact of a nanoparticle on model cell membranes is highly dependent on the presence of the soluble protein in the medium. We found that albumin boosts the activity of PAMAM against lipid bilayers that carry a net negative charge while no such effect is observed for membranes carrying zero net charge. These results are surprising given that albumin carries a net negative charge and the overall PAMAM–protein complexes are also negatively charged. However, the results can be rationalized in terms of the physical–chemical properties of the nanoparticle, protein and lipid system, where uneven charge distributions exist for the protein mainly giving it a dipolar character. This effect is also known as “charged patches” and can explain complexation between proteins and similarly charged polyelectrolytes (for a recent review see ref. 36). Such dipolar behavior can indeed fine-tune the overall activity of the PAMAM dendrimers against cell membranes.
Results
Impact of albumin on the interaction of PAMAM dendrimers with GUVs
To study the impact of BSA on the interaction of PAMAM G6 dendrimers with lipid membranes of different net charges we first employed Alexa Fluor® 488 hydrazide (Alexa 488)-loaded GUVs composed of different lipid compositions: pure POPC, a binary mixture of POPC and negatively charged POPG at a molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and a binary mixture of POPC and negatively charged POPS at a molar ratio of 3:1 using DiD-C18 as the membrane marker. Due to local concentration variations and GUV variability, GUV could appear clustered or could display smaller vesicles within even in the absence of BSA or PAMAM G6. In particular, this was often observed for uncharged vesicles (POPC). However, the vesicles did not display any change in fluorescence intensity or shape even up to 1 h of analysis excluding any significant photo-bleaching of the fluorescent probe and impact of mechanical stress upon pipetting (Fig. 1A, D and G). The addition of 0.5 μM PAMAM G6 dendrimers in the absence of BSA promoted leakage of the vesicles independent of their lipid composition (Fig. 1B, E and H) in line with previous data, and some vesicle aggregation for negative vesicles only.21 Notably, co-addition of BSA and PAMAM G6 dendrimers to negatively charged POPC/POPG vesicles resulted not only in leakage but simultaneously also in a more drastic vesicle aggregation (Fig. 1C) that was followed by collapse of several vesicles on the glass surface (ESI, Fig. S1†). Likewise, negatively charged vesicles composed of a binary mixture of POPC–POPS exhibited the same behaviour as observed with POPC/POPG vesicles (Fig. 1F), thereby ruling out that the observed effect results from the lipid headgroup structure of PG. Conversely, co-addition of BSA and PAMAM G6 dendrimers to GUVs composed of POPC caused leakage but the overall three-dimensional structure of the GUVs remained preserved (Fig. 1I).
:1, and a binary mixture of POPC and negatively charged POPS at a molar ratio of 3:1 using DiD-C18 as the membrane marker. Due to local concentration variations and GUV variability, GUV could appear clustered or could display smaller vesicles within even in the absence of BSA or PAMAM G6. In particular, this was often observed for uncharged vesicles (POPC). However, the vesicles did not display any change in fluorescence intensity or shape even up to 1 h of analysis excluding any significant photo-bleaching of the fluorescent probe and impact of mechanical stress upon pipetting (Fig. 1A, D and G). The addition of 0.5 μM PAMAM G6 dendrimers in the absence of BSA promoted leakage of the vesicles independent of their lipid composition (Fig. 1B, E and H) in line with previous data, and some vesicle aggregation for negative vesicles only.21 Notably, co-addition of BSA and PAMAM G6 dendrimers to negatively charged POPC/POPG vesicles resulted not only in leakage but simultaneously also in a more drastic vesicle aggregation (Fig. 1C) that was followed by collapse of several vesicles on the glass surface (ESI, Fig. S1†). Likewise, negatively charged vesicles composed of a binary mixture of POPC–POPS exhibited the same behaviour as observed with POPC/POPG vesicles (Fig. 1F), thereby ruling out that the observed effect results from the lipid headgroup structure of PG. Conversely, co-addition of BSA and PAMAM G6 dendrimers to GUVs composed of POPC caused leakage but the overall three-dimensional structure of the GUVs remained preserved (Fig. 1I).
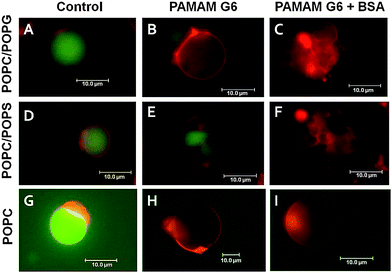 | ||
| Fig. 1 BSA enhances the impact of PAMAM G6 dendrimers on the integrity of negatively charged vesicles only. GUVs with three different lipid compositions were used: (A–C) POPC–POPG at a molar ratio of 75:25, (D–F) POPC–POPS at a molar ratio of 75:25, and (G-I) POPC only. DiD-C18 (red) and Alexa 488 (green) were used to visualize the membrane and the vesicle aqueous lumen, respectively. Vesicles were analyzed by fluorescence microscopy up to 1 h after addition of buffer (control: A, D and G), PAMAM G6 alone (B, E and H), or PAMAM G6 pre-incubated with BSA (C, F and I). For POPC–POPG and POPC/POPS vesicles the concentrations of PAMAM G6 and BSA were 0.5 μM and 5 μM, respectively. A 10 times higher BSA and PAMAM G6 concentration was used for pure POPC vesicles. BSA (5 μM) alone did not affect the vesicles (ESI, Fig. S2†). | ||
Control experiments in the absence of dendrimers revealed that BSA alone did not cause leakage or vesicle collapse over a broad concentration range regardless of the type of vesicle used (ESI, Fig. S2, and ref. 37). Moreover, content leakage occurred only for the negatively charged GUVs once the BSA concentration was raised to 10 μM while neutral vesicles were able to withstand up to 100 μM BSA without any significant leakage (ESI, Fig. S2, and ref. 37). We conclude that BSA enhances the effect of dendrimers on negatively charged vesicles only leading to the loss of membrane integrity with complete dye leakage, vesicle aggregation and vesicle collapse on the microscope slides.
Fig. 2 shows the total number of leakage events as a function of time for negative (A) and neutral vesicles (B) in the presence of BSA, PAMAM G6 or BSA–PAMAM G6 mixtures in the same concentrations as for Fig. 1. Since vesicles containing PG or PS behaved similarly in the presence of PAMAM dendrimers we decided to focus our work on POPC/POPG vesicles only. The changes reported correspond to several vesicles analyzed in a determined optical field, at six different positions of the microscope chamber. For negatively charged GUVs, the total number of leaked vesicles in the presence of BSA–G6 mixtures is significantly larger than the total number of leaked vesicles in the presence of BSA alone and dendrimers alone. This is though not the case for neutral vesicles for which the total number of leaked vesicles in the presence of BSA–G6 mixtures is equal to the total number of leaked vesicles in the presence of BSA alone and dendrimers alone. For neutral vesicles, the concentration of both dendrimers and BSA was raised by a factor of ten in order to see appreciable leakage events within the time of the experiment. Under these conditions, the extent of dye leakage from the lumen of the vesicles was within experimental error in the presence and absence of BSA.
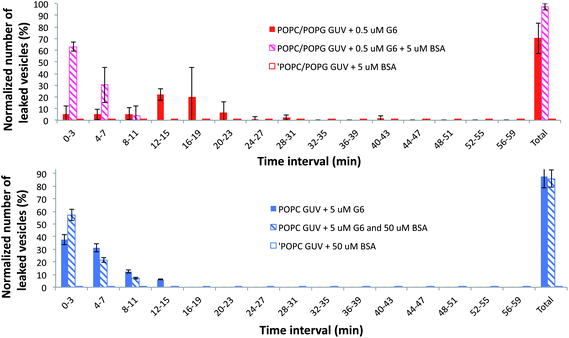 | ||
| Fig. 2 Leakage analysis of Alexa 488-loaded GUVs in the presence of PAMAM G6 and BSA. The upper figure gives representative histograms showing the normalized number of leaked POPC/POPG vesicles in time intervals of 3 min in the presence of 0.5 μM dendrimers and 5 μM BSA. The lower figure gives representative histograms showing the normalized number of leaked POPC vesicles in time intervals of 3 min in the presence of 5 μM dendrimers and 50 μM BSA. Data represent the mean of 3 independent experiments (with at least 30 vesicles analyzed at each experiment). Error bars show standard deviations. The lipid concentration was kept constant at ∼14 μM. The total number of leaked vesicles after 1 hour of visualization (total leakage) is also indicated in the figure. Similar results were found for GUVs made by electroswelling as indicated in Fig. S3 in the ESI,† indicating that the method of GUV formation has little impact on the permeability of lipid membranes upon interaction with dendrimers. | ||
Moreover, the rate at which each individual vesicle leaked was faster when PAMAM was added in the presence of BSA as exemplified in Fig. 3. For BSA (upper row), no significant change in fluorescence signal is observed within the time of the experiment. This confirms that no significant photobleaching occurs within the experimental time. For the PAMAM G6–BSA mixture (middle row), complete leakage occurs within the first 5 min and the vesicle collapses and moves out of the confocal volume (see Fig. S1 in the ESI† for an image of the confocal plane on the surface where the collapsed vesicle is clearly visible). For PAMAM G6 only (lower row), the leakage kinetics seems to have two regimes: (1) 60% of the fluorescence intensity is lost within the initial 5 min while (2) the remaining ∼40% intensity is lost during the next 40 min. Some of the original vesicles contained smaller vesicles within or in close adjacency to them although no obvious vesicles were observed in the confocal plane shown in Fig. 3 (first column). The slowing down of the leakage for vesicles exposed to dendrimers alone could be due to the fluorescence signal of trapped vesicles that diffuse in the confocal volume during the time of the experiment. Another alternative is that the lipid bilayers initially perturbed by dendrimer adsorption slowly rearrange filling back at least partially the regions of lower lipid density that – probably – are behind the increased permeability of the lipid bilayer towards the soluble dye.
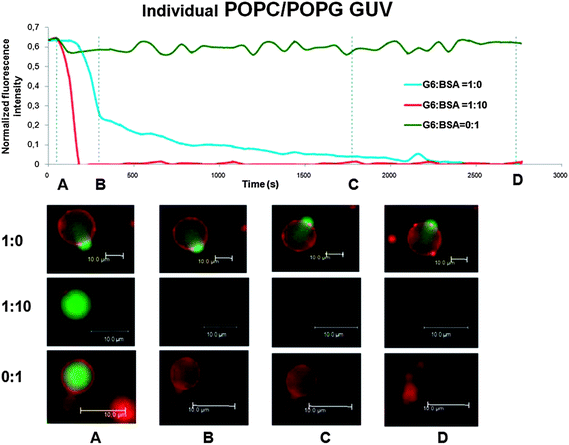 | ||
| Fig. 3 Fluorescence intensity traces versus elapsed time for leaking vesicles upon addition of 0.5 μM dendrimers in the absence and presence of BSA (5 μM) for POPC/POPG vesicles. The figure also includes the control signal for GUVs exposed to 5 μM BSA only. The background (0.35 a.u.) was subtracted from the signal and the change in fluorescence intensity was normalized to the fluorescence intensity before exposure to PAMAM G6 or PAMAM G6–BSA. Distinctive confocal images for each set of experiments are shown for the time frames marked by the broken line. The vesicle membrane was imaged with DiD-C18 (red) while the vesicle aqueous lumen was imaged with Alexa 488 (green). | ||
In order to analyze directly the interaction of PAMAM G6 dendrimers with vesicles in the absence and presence of BSA, FITC-labeled dendrimers were used during imaging (Fig. 4). To ensure a minimal modification of the PAMAM G6 dendrimers by the FITC labeling, only ∼1% of the dendrimer terminal groups were linked to FITC. In this set of experiments, Atto 550 instead of Alexa 488 was used to label the vesicle aqueous lumen. Analysis by confocal scanning fluorescence microscopy revealed intense dendrimer accumulation on the surface of the negatively charged POPC/POPG vesicles in the absence and presence of BSA (Fig. 3). These images focus on the vesicle aggregation stage occurring prior to their collapse. A significantly more intense signal was consistently observed for FITC-labeled dendrimers around POPG-containing membranes in the presence of BSA. Under the same conditions, no significant dendrimer–lipid interaction occurred for POPC vesicles regardless of the presence of BSA, instead the labeled dendrimers preferentially interacted with the BSA coated microscope slide (ESI, Fig. S4†). Similar results were obtained for a more physiologically relevant salt concentration, i.e. 150 mM NaCl (ESI, Fig. S5†). Interestingly, Fig. 4 shows that vesicles seem to exhibit different FITC intensities within the same experiment thus implying different dendrimer concentrations around the vesicles. The relative intensities do not seem to be related to the size of the vesicles and could instead be related to heterogeneities in the lipid composition at the single vesicle level. Similar heterogeneities at the single vesicle level have been reported earlier by fluorescence microscopy38 and by atomic force microscopy.32
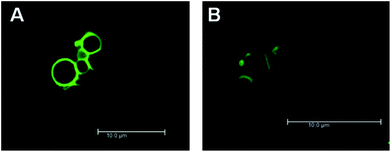 | ||
| Fig. 4 BSA boosts PAMAM G6 accumulation on the surface of negatively charged vesicles. Atto 550-loaded vesicles prepared from POPC–POPG mixtures (75:25, molar ratio) were incubated with 0.5 μM FITC-labeled PAMAM G6 (green) in the presence (A) or absence (B) of 5 μM BSA. The lipid concentration was kept constant at ∼14 μM. Images were recorded after lumen leakage and prior to vesicle collapse. | ||
PAMAM G6 forms complexes with BSA
Complex formation of G6 PAMAM dendrimer with serum albumin protein has been thoroughly documented. However, previous work focuses on the effect of dendrimers on the protein structure29,39–41 and only one recent report focuses on fibrinogen aggregation due to dendrimers addition to blood.33 In order to confirm whether cationic dendrimers self-assemble with BSA to form stable complexes under our experimental conditions we analyzed solutions of 150 μM BSA, 45 μM PAMAM G6 and their mixtures (1:10 molar ratio) by dynamic light scattering (DLS), a technique providing the apparent size distribution of complexes. Results from DLS measurements are given in Fig. 5A. BSA displays a single diffusional mode with an apparent hydrodynamic radius (RappH) of ∼4 nm while PAMAM G6 shows a diffusional mode also at ∼4 nm and a non-diffusional mode at ∼71 nm which is typical for polyelectrolytes.42 This non-diffusional mode typically arises from the coupled motion of the polyelectrolyte and its counterions as discussed by Ainelem et al.42 The BSA–PAMAM G6 mixture also showed two modes, one at ∼4 nm and another at 270 nm. The latter mode corresponds to large BSA–dendrimer aggregates. Similar DLS results were obtained at 1:1 and 1:5 PAMAM G6:BSA molar ratios.
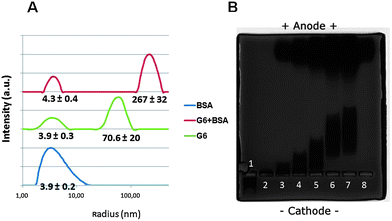 | ||
| Fig. 5 Physical–chemical behavior of BSA–G6 complexes. (A) Dynamic light scattering measurements for BSA (150 μM), G6 (40 μM) and G6–BSA mixtures 1:10 (15 μM BSA + 1.5 μM PAMAM G6). Upon mixing dendrimers with BSA, large aggregates are formed. The indicated values for the apparent hydrodynamic radii (RappH) represent mean ± SD for 4–6 independent measurements. (B) Agarose gel electrophoresis for 9 μM PAMAM G6 dendrimers upon increasing concentrations of BSA. Samples from left to right consist of a PAMAM G6 to BSA molar ratio of 1:0 (lane 1), 1:1.5 (lane 2), 1:8 (lane 3), 1:16 (lane 4), 1:33 (lane 5), 1:66 (lane 6) and 1:100 (lane 7), while lane 8 corresponds to 75 μM BSA without dendrimers. Complexes of dendrimer and proteins stay close on the wells at the cathode side, while unbound proteins show a larger migration. | ||
Complex formation between dendrimers and BSA was further confirmed by gel electrophoresis (Fig. 5B). PAMAM G6 dendrimers have 256 terminal amino groups (ESI, Table S1†) and carried a positive net charge under the ionic conditions used in this work (zeta potential, 16.9 ± 0.5 mV). As expected, cationic dendrimers did not migrate towards the anode under these conditions but instead moved towards the cathode (lane 1). At pH 7.4, BSA carries a net negative charge (see Table S1†) under the ionic conditions used in this work (zeta potential, −11.4 ± 0.9 mV) and thus unbound proteins had a significant higher migration velocity and moved to the anodal front (lane 8). For dendrimers pre-incubated with BSA migration towards the anode occurred, indicating the formation of complexes with a net negative charge (lanes 2–7). Increased migration occurred for the dendrimer incubated with higher concentration of BSA and no saturation in terms of migration distance was observed with BSA concentrations up to the protein to dendrimer molar ratio of 100. These data indicate that the charge of BSA–dendrimer complexes increases with the BSA concentration. Taken together, the migration data and DLS results suggest that complexes between PAMAM G6 dendrimers and BSA are probably extended and/or grafted aggregates. Indeed, based on the zeta potential values of BSA (−11.4 ± 0.9 mV) and PAMAM G6 (16.9 ± 0.5 mV), more than one BSA should be able to bind each dendrimer molecule. Note that the size of the migrating aggregates must be at least smaller than 200 nm, the mesh size of the agarose gel.43 This mesh size is in the same range as RappH determined by DLS. However, RappH is calculated using the Stokes–Einstein equation and thus assuming a spherical shape. The actual shape of these aggregates may be indeed non-spherical and thus the DLS values can only be used as rough estimates of the actual size. Since it was not possible to separate the PAMAM–BSA complexes from the unbound BSA molecules, we could not use other complementary characterization methods such as zeta potential measurements for determination of the actual charge of the PAMAM G6–BSA aggregates.
Discussion
Previously, we demonstrated that PAMAM G6 dendrimers affected the structure of giant unilamellar vesicles (GUVs),21 small unilamellar vesicles (SUVs)20 and supported lipid bilayers (SLBs).22 The dendrimers induced the collapse of the vesicles on the microscope slides once a certain critical surface concentration was reached. Such vesicle collapse compares with the morphological changes occurring in red blood cells after exposure to high dendrimer concentrations24 and appears to be induced by the stiffening of the cell membrane.21 In this work we show that BSA exacerbates the effect of PAMAM G6 on the destabilization of the vesicles in terms of leakage, aggregation and collapse for negatively charged vesicles (Fig. 1–3). DLS and agarose gel electrophoresis revealed extended complex formation between PAMAM G6 and BSA (Fig. 5). Although the complexes carry a net negative charge, PAMAM–BSA complexes are very active towards other negative surfaces. Surprisingly, the PAMAM G6 complexes seem to display a higher affinity for the negatively charged lipid vesicles than the pristine dendrimers since a denser PAMAM G6 layer formed around the surface of the vesicles in the presence of BSA (Fig. 4). Notably, no such synergy occurred for neutrally charged vesicles (Fig. 2). Instead, labeled dendrimers preferentially adsorbed on the BSA surface (Fig. SI4†).BSA itself has certain affinity for negatively charged vesicles in the absence of PAMAM G6 dendrimers. Content leakage from POPG-containing vesicles occurred at 10 μM BSA while the same effect was not observed for neutral vesicles at least up to 100 μM BSA (ESI, Fig. S2† and ref. 37). The crystal structure of BSA44 reveals that the protein’s surface is a highly contoured landscape with an uneven and distinctive charge distribution. Most negative charges are located in domains I and II whereas the positive charges are mainly distributed in domain III. Thus, this protein presents a permanent charge dipole that can lead to attractive electrostatic interactions between the regions containing higher density of negatively charged groups and any positively charged particles such as PAMAM G6. This characteristic of proteins is typically known as “charged patches” (for a recent review see ref. 36) and explains the binding of proteins and similarly charged polyelectrolytes. The more positively charged enriched domain (III) is then able to strongly interact with negative charged objects45 such as the surface of POPC/POPG vesicles. This “dipole” character of BSA may lead to the formation of extended mixed BSA–PAMAM G6 complexes as schematically represented in Fig. 6. The local surface charge of these extended PAMAM G6–BSA complexes is thus considerably higher than for PAMAM only since not only PAMAM but also BSA contribute to the electrostatic interaction with the charged lipid surface, rendering the complexes more active against these vesicles. There is moreover an entropic contribution upon BSA–lipid membrane binding due to the release of bound counterions to the bulk solution.46 On the other hand, BSA adsorption occurred on the negatively charged vesicles in BSA concentrations at which no effect on the membrane's permeability occurred (1 μM).37 Thus, BSA adsorption should also occur in our case and the observed synergism could be due in part to additional perturbation of the lipid packing by BSA adsorption on the lipid membrane. Moreover, QCM-D measurements37 showed that BSA adsorption is only partially reversible on negative lipid membranes. This could be due to partial intercalation in the hydrophobic region of the lipid bilayer thus facilitating the destabilizing effect of dendrimers and dendrimer–BSA complexes on the lipid vesicles. However, the G6 effect on the permeability of GUVs that have been pre-exposed to BSA for 15 min and subjected to 3 volume washes with PBS was similar to the effect for vesicles exposed to G6 in the absence of BSA (ESI, Fig. S3†). Thus, the effect of BSA on the membrane cannot account for the increased permeability of POPC/POPG vesicles in the presence of G6–BSA complexes rather the observed synergism must be a direct consequence of the PAMAM G6–BSA complex adsorption on the vesicle surface.
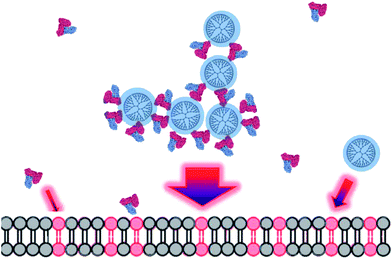 | ||
| Fig. 6 A model for PAMAM G6 and PAMAM G6–BSA complexes interacting on the surface of negatively charged GUVs. The cartoon shows dendrimer–BSA aggregates with a branched necklace structure approaching a POPC–POPG lipid bilayer. Positively charged regions are colored in blue, while negatively charged ones are presented in red; neutral ones are in black. Here the negative domains of the protein are supposed to link the dendrimers and drive them towards the negative lipids, where domain III is located outwards in this complex due to its positive charge. The BSA crystal structure (Protein Data Bank accession codes 3V03) was taken from ref. 44. The size of the arrows illustrates the different relative affinities of the molecules and complexes formed by BSA and PAMAM molecules. The protein and dendrimer are drawn to scale while the lipid bilayer is out of scale. | ||
Our experiments show that even at a very low dendrimer concentration of 0.5 μM, the presence of BSA boosts the activity of PAMAM G6 against negatively charged vesicles. On the other hand, no synergism occurred in the presence of BSA for neutral vesicles. Even though large complexes of BSA and PAMAM also were present in this case, the electrostatically driven synergism between BSA and PAMAM aggregates no longer was valid. In this case, BSA did not have any attractive interaction with the neutral vesicles as evidenced by the ability of POPC vesicles to withstand up to 100 μM BSA without any apparent effect on its structure. Moreover, it is clear that dendrimers have a higher affinity for BSA than neutrally charged lipid bilayers as demonstrated by the preferential adsorption of FITC labeled dendrimers on the BSA coated surface (see ESI, Fig. SI4†). Thus, overall the PAMAM G6–BSA aggregates present a lower affinity for neutrally charged vesicles.
PAMAM dendrimers are considered to have high antibacterial activity and low cytotoxicity. Besides other major structural differences, the bacterial cell membrane is known to display more negative charged lipids on the cell surface than mammalian cells, especially red blood cells.47–49 Thus, it is clear that the difference in net charge between various cell membranes might have a major impact on the activity/toxicity of PAMAM dendrimers. This effect would be even more evident in the presence of albumin or other soluble proteins with similar structure since in this case PAMAM dendrimers could interact with the bacteria surface even at very low concentrations due to the higher affinity of the PAMAM–protein complexes towards negative surfaces. At the same time, the protective effect of BSA observed for PAMAM on red blood cells10 may actually be caused by a reduction of the effective free dendrimer concentration as aggregates (with lower affinity for the membrane than pristine PAMAM molecules) are formed given that a low total dendrimer concentration was used in the assays.
In spite of the simplified experimental conditions in our experimental setup, the model lipid bilayer here employed seems to reproduce results observed in cell cultures. This is to a certain extent surprising given that the living cell membrane is stiffer, more resistant and can adjust its surface tension in order to defend itself from the medium outside, all mechanisms that are absent for the giant vesicles. Furthermore, our results appear in agreement with previous work in which cooperativity between histones and similarly charged polyethylenimine polymers occurred for the compaction and transfection of DNA.50 In particular, preferential interaction theory and molecular dynamics simulations were used to show that additional hydrogen bonding between L-arginine, L-glutamic acid and Drosophilia Su(dx) protein (ww34) led to enhanced amino acid crowding around the protein, suppressing the inter-protein association and increasing the protein solubility. Similarly, in our case the role of the inter-additive (albumin) interactions determines the extent of dendrimer–lipid interactions.
Conclusions
Our experiments clearly show that albumin acts as a modulator of the interactions between PAMAM G6 dendrimers and model cell membranes and this modulation is highly dependent on the net charge of the lipid membrane. There was a synergistic effect on negative charged giant unilamellar vesicles while no such effect occurred towards neutrally charged vesicles. These results put in evidence the delicate equilibrium between competing interactions for multicomponent systems, where a change in the affinities between two components (BSA and the lipid bilayer) may or may not boost the dendrimer effect on the lipid bilayer structure.Experimental section
Materials
POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-1′-rac-glycerol), POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine), POPS (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine) and DOPE–biotin (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-cap biotinyl) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) and were used without further purification. The fluorescent dyes DiDC18 (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanineperchlorate) and Alexa 488 (Alexa Fluor® 488 hydrazide) were likewise used as received from Invitrogen (Paisley, UK); Atto 550 was purchased from Fluka Analytical. Unless otherwise stated, all other chemicals and reagents were purchased from Sigma-Aldrich A/S (Copenhagen, Denmark). Milli-Q quality water was used in all preparations. Physical–chemical properties of the components used are summarized in the ESI, Table S1.†Protein and dendrimer solutions
Bovine serum albumin (BSA, purity >98%) was dissolved at a concentration of 1 mM in phosphate buffer saline (PBS, pH 7.4 containing 100 mM NaCl unless otherwise stated) and diluted further to the desired experimental concentration at the beginning of the experiments. PAMAM G6 dendrimers ([NH2(CH2)2NH2]:(G = 6); dendrimer PAMAM(NH2)256) were subjected to vacuum for complete evaporation of methanol before dispersion in PBS.
Dendrimer labeling
Fluorescein isothiocyanate (FITC) was covalently conjugated to the amine groups of the dendrimers through the formation of thiourea bonds as described elsewhere.21 Briefly, FITC dissolved in methanol was slowly added to the dendrimer solution in a molar ratio of 1:5 dendrimer:FITC. Unreacted FITC was removed by dialysis against PBS buffer for 3 days or until no free dye was observed in the dialysis buffer. Attachment of FITC to dendrimers was verified by spectrofluorimetry (excitation at 475 nm; emission at 583 nm) using a Fluoromax-4 (Horiba, Edison New Jersey, USA) as described earlier.21 An average molar ratio of 2.8 FITC molecules per dendrimer was obtained (each G6 dendrimer has 256 terminal groups).
GUV preparation
GUVs were prepared by gentle hydration of lipid films according to previous protocols.21 Briefly, lipids in chloroform were mixed in glass tubes to achieve the indicated lipid composition. Additionally, 1 mol% DiD-C18 and 0.5 mol% DOPE–biotin were added to the bulk lipids. Then, a thin lipid film was prepared by drop-by-drop addition of the mixture to small Teflon cups. The remaining chloroform was removed by storing the Teflon cups in a vacuum-chamber for 1 h. Lipids were rehydrated in D-sorbitol solution (46.1 g L−1 in PBS) supplemented with 10 μM soluble dye to a lipid concentration of 0.5 mg mL−1. After one night of incubation at 37 °C all vesicles were stored at 4 °C for a maximum of one week before use. Table S2† in the ESI summarizes the properties and applications of the dyes used in this work.For GUV preparation by electroswelling, a commercially available Vesicle Prep Pro® system was used at a 0.5 g L−1lipid concentration using the same sorbitol solution as used for the hydration method. An alternating voltage (3 V) with a frequency of 5 Hz was applied for 120 min.
Surface functionalization of the microscope chambers
Glass coverslips were cleaned carefully by successive washes in Helmanex (2%), Milli-Q water and ethanol. For surface functionalization, 1 g L−1 BSA–biotin:BSA (1:10, weight ratio) was added to the surface and incubated at ambient temperature for 10 min. After five times gentle washing with PBS, streptavidin (0.025 g L−1 in PBS) was added and likewise incubated for 10 min followed by five times washing with PBS. After surface functionalization, GUVs were added to the microscope chamber to a final constant lipid concentration of 0.01 g L−1 (∼14 μM) and allowed to stabilize for at least 30 min before the measurement. The total volume of the microscope chamber was adjusted to ∼200 μL. Dendrimers, BSA and their mixtures (premixed 30 min prior to the addition to the chamber) were pipetted in aliquots of 20 μL.
Fluorescence microscopy
Microscopy imaging was performed on an inverted confocal microscope TCS SP5 (Leica, Wetzlar, Germany) and a wide field microscope AF6000LX (Leica). In the confocal microscope, Alexa 488 and FITC-labeled dendrimers were excited with a 488 nm argon laser and emission was recorded between 491 and 563 nm. DiD-C18 was excited with a 633 nm laser and the emission was recorded between 640 and 700 nm. Atto550 was excited with a 556 nm laser and emission was recorded between 576 and 578 nm. In the wide-field microscope a mercury lamp with EGF 49002 ET and EC5 49006 ET filter cubes (Chroma Technology Corp, Bellows Falls, USA) was used to excite Alexa 488/FITC and DiD18, respectively. During wide-field microscopy experiments the illumination intensity and exposure time were changed for the experimental setup to maximize the signal. For confocal microscopy observations the signal was maximized setting the argon laser intensity to 19%, the acousto-Optic Tunable Filter for Attota to 21% and the photomultiplier voltage to 537.4 V. These settings were kept fixed during the various experiments in order to be able to perform a fluorescence intensity comparison study. The integrity of vesicles after the addition of buffer solution was checked during control experiments in order to ensure their stability against mechanical stress.Dynamic Light Scattering (DLS)
An ALV-5000 goniometer setup (ALV-GmbH, Langen, Germany) was used for DLS measurements at 90°. The light source was a 633 nm diode pumped Nd:YAG solid-state Compass-DPSS laser (COHERENT, Inc., Santa Clara, CA). The temperature was controlled at 25 ± 0.1 °C. The hydrodynamic radius was obtained from the study of the autocorrelation function of the laser signal scattered from the sample using the Stocks–Einstein equation, as described elsewhere (see for example ref. 51). The hydrodynamic radii presented are averages from 4–6 measurements.Agarose gel electrophoresis
The binding ability of PAMAM G6 towards BSA was examined by the gel retardation assay. Four volumes of samples were mixed with one volume of non-denaturing loading buffer (8 mM Tris–HCl, 0.2 mM EDTA, 50% glycerol, 0.01% (v/w) bromphenol blue) and equal volumes were loaded on 0.5% agarose gel. Gel electrophoresis was performed at 130 V in standard Tris–acetate–EDTA buffer for at least 3 h for optimal separation. To prevent heating of the gel, equipment was placed in an ice water bath during the separation and the buffer was exchanged at least once every hour. Straight after electrophoresis, the gel was stained with Coomassie Brilliant Blue R-250.Acknowledgements
Confocal Imaging data were collected at the Center for Advanced Bioimaging (CAB) Denmark, University of Copenhagen. We thank Karen Martinez for access to the wide field microscope. Moreover, the authors gratefully acknowledge financial support from the “Center for Synthetic Biology” at Copenhagen University funded by the UNIK research initiative of the Danish Ministry of Science, Technology and Innovation.References
- R. Esfand and D. A. Tomalia, Drug Discovery Today, 2001, 6, 427–436 CrossRef CAS.
- O. A. Matthews, Prog. Polym. Sci., 1998, 23, 1–56 CrossRef CAS.
- A. K. Patri, J. F. Kukowska-Latallo and J. R. Baker, Jr, Adv. Drug Delivery Rev., 2005, 57, 2203–2214 CrossRef CAS.
- A. Quintana, E. Raczka, L. Piehler, I. Lee, A. Myc, I. Majoros, A. K. Patri, T. Thomas, J. Mule and J. R. Baker, Jr, Pharm. Res., 2002, 19, 1310–1316 CrossRef CAS.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS.
- J. A. Barreto, W. O'Malley, M. Kubeil, B. Graham, H. Stephan and L. Spiccia, Adv. Mater., 2011, 23, H18–H40 CrossRef CAS.
- B. Pan, D. Cui, P. Xu, C. Ozkan, G. Feng, M. Ozkan, T. Huang, B. Chu, Q. Li, R. He and G. Hu, Nanotechnology, 2009, 20, 125101 CrossRef.
- H. Kobayashi and M. W. Brechbiel, Mol. Imaging, 2003, 2, 1–10 CrossRef CAS.
- A. Mecke, I. J. Majoros, A. K. Patri, J. R. Baker, M. M. B. Holl and B. G. Orr, Langmuir, 2005, 21, 10348–10354 CrossRef CAS.
- A. Mecke, S. Uppuluri, T. M. Sassanella, D.-K. Lee, A. Ramamoorthy, J. R. Baker Jr, B. G. Orr and M. M. Banaszak Holl, Chem. Phys. Lipids, 2004, 132, 3–14 CrossRef CAS.
- S. Parimi, T. J. Barnes and C. A. Prestidge, Langmuir, 2008, 24, 13532–13539 CrossRef CAS.
- S. Hong, A. U. Bielinska, A. Mecke, B. Keszler, J. L. Beals, X. Shi, L. Balogh, B. G. Orr, J. R. Baker and M. M. Banaszak Holl, Bioconjugate Chem., 2004, 15, 774–782 CrossRef CAS.
- S. Hong, P. R. Leroueil, E. K. Janus, J. L. Peters, M.-M. Kober, M. T. Islam, B. G. Orr, J. R. Baker and M. M. Banaszak Holl, Bioconjugate Chem., 2006, 17, 728–734 CrossRef CAS.
- M. F. Ottaviani, S. Jockusch, N. J. Turro, D. A. Tomalia and A. Barbon, Langmuir, 2004, 20, 10238–10245 CrossRef CAS.
- M. F. Ottaviani, P. Matteini, M. Brustolon, N. J. Turro, S. Jockusch and D. A. Tomalia, J. Phys. Chem. B, 1998, 102, 6029–6039 CrossRef CAS.
- C. V. Kelly, P. R. Leroueil, B. G. Orr, M. M. Banaszak Holl and I. Andricioaei, J. Phys. Chem. B, 2008, 112, 9346–9353 CrossRef CAS.
- K. Gardikis, S. Hatziantoniou, K. Viras, M. Wagner and C. Demetzos, Int. J. Pharm., 2006, 318, 118–123 CrossRef CAS.
- M.-L. Ainelem, R. A. Campbell, S. Khalid, R. J. Gillams, A. R. Rennie and T. Nylander, J. Phys. Chem. B, 2010, 114, 7229–7244 CrossRef.
- S. Parimi, T. J. Barnes and C. A. Prestidge, Langmuir, 2008, 24, 13532–13539 CrossRef CAS.
- A. Åkesson, K. M. Bendtsen, M. A. Beherens, J. S. Pedersen, V. Alfredsson and M. Cárdenas Gómez, Phys. Chem. Chem. Phys., 2010, 12, 12267–12272 RSC.
- A. Åkesson, C. V. Lundgaard, N. Ehrlich, T. G. Pomorski, D. Stamou and M. Cárdenas, Soft Matter, 2012, 8, 8972–8980 RSC.
- A. Åkesson, T. Lind, R. Barker, A. Hughes and M. Cárdenas, Langmuir, 2012, 28, 13025–13031 CrossRef.
- B. Klajnert, S. Pikala and M. Bryszewska, Proc. R. Soc. London, Ser. A, 2010, 466, 1527–1534 CrossRef CAS.
- N. Malik, R. Wiwattanapatapee, R. Klopsch, K. Lorenz, H. Frey, J. W. Weener, E. W. Meijer, W. Paulus and R. Duncan, J. Controlled Release, 2000, 65, 133–148 CrossRef CAS.
- M. El-Sayed, M. Ginski, C. Rhodes and H. Ghandehari, J. Controlled Release, 2002, 81, 355–365 CrossRef CAS.
- K. M. Kitchens, R. B. Kolhatkar, P. W. Swaan, N. D. Eddington and H. Ghandehari, Pharm. Res., 2006, 23, 2818–2826 CrossRef CAS.
- J. Giri, M. S. Diallo, A. J. Simpson, Y. Liu, W. A. Goddard, R. Kumar and G. C. Woods, ACS Nano, 2011, 5, 3456–3468 CrossRef CAS.
- D. Shcharbin, B. Klajnert and M. Bryszewska, J. Biomater. Sci., Polym. Ed., 2005, 16, 1081–1093 CrossRef CAS.
- E. Froehlich, J. S. Mandeville, C. J. Jennings, R. Sedaghat-Herati and H. A. Tajmir-Riahi, J. Phys. Chem. B, 2009, 113, 6986–6993 CrossRef CAS.
- B. Klajnert and M. Bryszewska, Bioelectrochemistry, 2002, 55, 33–35 CrossRef CAS.
- E. Gabellieri, G. B. Strambini, D. Shcharbin, B. Klajnert and M. Bryszewska, Biochim. Biophys. Acta, 2006, 1764, 1750–1756 CrossRef CAS.
- A. Åkesson, M. Cárdenas, G. Elia, M. P. Monopoli and K. A. Dawson, RSC Adv., 2012, 2, 11245 RSC.
- C. F. Jones, R. A. Campbell, A. E. Brooks, S. Assemi, S. Tadjiki, G. Thiagarajan, C. Mulcock, A. S. Weyrich, B. D. Brooks, H. Ghandehari and D. W. Grainger, ACS Nano, 2012, 6, 9900–9910 CrossRef CAS.
- C. Plank, K. Mechtler, F. C. Szoka and E. Wagner, Hum. Gene Ther., 1996, 7, 1437–1446 CrossRef CAS.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS.
- A. B. Kayitmazer, D. Seeman, B. B. Minsky, P. L. Dubin and Y. Xu, Soft Matter, 2013, 9, 2553 RSC.
- F. Ruggeri, F. Zhang, T. Lind, E. D. Bruce, B. L. T. Lau and M. Cárdenas, Soft Matter, 2013, 9, 4219–4226 RSC.
- J. Larsen, N. S. Hatzakis and D. Stamou, J. Am. Chem. Soc., 2011, 133, 10685–10687 CrossRef CAS.
- D. Shcharbin, B. Klajnert, V. Mazhul and M. Bryszewska, J. Fluoresc., 2005, 15, 21–28 CrossRef CAS.
- D. Shcharbin, M. F. Ottaviani, M. Cangiotti, M. Przybyszewska, M. Zaborski and M. Bryszewska, Colloids Surf., B, 2008, 63, 27–33 CrossRef CAS.
- B. Klajnert, L. Stanislawska, M. Bryszewska and B. Palecz, Biochim. Biophys. Acta, 2003, 1648, 115–126 CrossRef CAS.
- M.-L. Ainelem, A. M. Carnerup, J. Janiak, V. Alfredsson, T. Nylander and K. Schillén, Soft Matter, 2009, 5, 2310–2320 RSC.
- B. J. Smith, in Proteins, 1984, ch. 6, vol. 1, pp. 41–55 Search PubMed.
- Q. Shi, Y. Zhou and Y. Sun, Biotechnol. Prog., 2005, 21, 516–523 CrossRef CAS.
- B. Jachimska and A. Pajor, Bioelectrochemistry, 2012, 87, 138–146 CrossRef CAS.
- C. Wang and K. C. Tam, Langmuir, 2002, 18, 6484–6490 CrossRef CAS.
- M. V. Lizenko, T. I. Regerand, A. M. Bakhirev and E. I. Lizenko, J. Evol. Biochem. Physiol., 2011, 47, 428–437 CrossRef CAS.
- G. van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef CAS.
- R. M. Epand and R. F. Epand, Mol. BioSyst., 2009, 5, 580–587 RSC.
- A. Schneeweiss, K. Buyens, M. Giese, N. Sanders and S. Ulbert, Int. J. Pharm., 2010, 400, 86–95 CrossRef CAS.
- M. Cárdenas, K. Schillén, T. Nylander, J. Jansson and B. Lindman, Phys. Chem. Chem. Phys., 2004, 6, 1603 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Typical images of vesicle collapse on the microscope slides, the effect of BSA on GUVs, confocal images of the dendrimer interaction with POPC vesicles, and the effect of using higher ionic strength in the assay. See DOI: 10.1039/c3sm50603g |
| This journal is © The Royal Society of Chemistry 2013 |