 Open Access Article
Open Access ArticleSelf-assembly of a model amphiphilic oligopeptide incorporating an arginine headgroup†
Ian W. Hamley*a, Ashkan Dehsorkhia, Valeria Castellettoa, Jani Seitsonenb, Janne Ruokolainenb and Hermis Iatrouc
aDepartment of Chemistry, University of Reading, Reading, RG6 6AD, UK. E-mail: I.W.Hamley@reading.ac.uk
bDepartment of Applied Physics, Aalto University School of Science, P.O.Box 15100, FI-00076 Aalto, Finland
cUniversity of Athens, Department of Chemistry, Panepistimiopolis Zografou, 157 71 Athens, Greece
First published on 28th March 2013
Abstract
The self-assembly in aqueous solution of the alanine-rich peptide A12R2 containing twelve alanine residues and two arginine residues has been investigated. This oligomeric peptide was synthesized via NCA-polymerization methods. The surfactant-like peptide is found via FTIR to form antiparallel dimers which aggregate into twisted fibrils, as revealed by cryogenic-transmission electron microscopy. The fibril substructure is probed via detailed X-ray scattering experiments, and are uniquely comprised of twisted tapes only 5 nm wide, set by the width of the antiparallel A12R2 dimers. The packing of the alanine residues leads to distinct “β-sheet” spacings compared to those for amyloid-forming peptides. For this peptide, β-sheet structure coexists with some α-helical content. These ultrafine amyloid fibrils present arginine at high density on their surfaces, and this may lead to applications in nanobiotechnology.
Introduction
Amphiphilic peptides combine the biofunctionality and bioactivity afforded by sequences of amino acids with amphiphilicity. Surfactant-like peptides1,2 are a class of amphiphilic peptide comprising a headgroup which is a short sequence of charged residues attached to a tailgroup of neutral residues. Examples include A6D, V6D, V6D2, L6D2 G4D2, V6K2, A6K, etc.3,4 Zhang and coworkers have shown that these materials have outstanding potential in the development of novel biomaterials.5 For example, they can be used to stabilize the conformation of proteins in solution, such as G protein-coupled receptor (GPCR) bovine rhodopsin.6 More recently, this work was developed to exploit peptide surfactants in the production and stabilization of membrane proteins such as GPCRs.7In previous work, we have examined the self-assembly of the surfactant-like peptide A6K (prepared as TFA salt), introduced by Zhang et al.,8,9 into nanotubes.10 Flow-aligning X-ray diffraction enabled a detailed model for the helical arrangement of the peptides within the walls of the nanotubes to be proposed. This was based on salt-bridged dimers, with a two-residue offset shift between adjacent dimers.10 This research built on earlier work by Bucak et al. which indicated that, at sufficiently high concentration in aqueous solution A6K forms nanotubes with a very thin wall, possibly single wall nanotubes, stabilized by the headgroup charge.11
In the present work, we investigate the self-assembly, of A12R2, containing two arginine residue as headgroup. Arginine is a highly basic residue (pKa = 12 approximately12) incorporating a guanidinium group which due to delocalization of the charge is able to form bidentate hydrogen bonds.13 Cell-penetrating peptides such as the TAT protein transduction domain14 peptide or penetratin15 are often rich in arginine residues16 and are extensively used as potential delivery vehicles for siRNA and DNA. Compared to most cell-penetrating peptides A12R2 presents a greatly simplified surfactant-like design and in the present paper we examine its self-assembly behavior which turns out to have a number of remarkable features. The oligomeric peptide is synthesized using N-carboxyanhydride (NCA) polymerization which is a valuable technique for the synthesis of “block copolypeptides”.17 Standard solid phase synthesis methods would be extremely problematic for peptides of this type due to the multiple repeat units, which would lead to aggregation during synthesis causing the growing chain to fold back on itself, meaning that the incoming amino acid cannot be added causing chain termination usually between a 7-mer and 9-mer. This could be disrupted by placing aggregation disrupting units (“kinks”) in the chain, however this would disrupt the sequence. These problems are avoided using the NCA technique. We show here that A12R2 self-assembles into unprecedented ultrathin amyloid fibrils, with a width set by the length of the molecule, and a fundamental antiparallel dimer building unit.
Experimental
Materials
The polymerization solvent, DMF (Aldrich, 99.9+%) was a special grade for peptide synthesis (active impurities <50 ppm) and was further purified by short-path fractional distillation on the vacuum line at low temperature under high vacuum, in a custom-made apparatus. Only the middle fraction was used. Ethyl acetate was dried over phosphorus pentoxide and fractionally distilled under high vacuum. n-Hexane was dried overnight over CaH2 and then distilled into a flask containing n-BuLi. Acetonitrile (Aldrich 99.9%) was dried over phosphorus pentoxide and then fractionally distilled prior to use. Dimethylamine, the initiator, was condensed in a flask containing sodium mirror at −10 °C and was then diluted in purified DMF and subdivided into ampoules. Besides the monomer synthesis, the purifications and polymerizations were performed using high vacuum techniques.18Synthesis of N-carboxy anhydrides (NCAs)
The synthesis of the alanine NCA (Ala-NCA) has already been presented in a previous work.19 Briefly, Ala-NCA was synthesized from the corresponding L-amino acid and triphosgene in acetonitrile (suspension) at 70 °C, under inert atmosphere. The unreacted species along with the amino acid salts (insoluble species) were removed by filtration. Ala-NCA was subsequently dissolved and dried several times with ethyl acetate under high vacuum, in order to remove the excess triphosgene, which sublimes under high vacuum, along with the remaining HCl. Finally, Ala-NCA was dissolved in ethyl acetate, and was recrystallized from n-hexane three times under high vacuum. The purified NCA was stored under an inert atmosphere at 0 °C.The NCA of N-delta,N-omega-di-(benzyloxycarbonyl)-L-arginine (L-Arg(Di-Z)-NCA) was synthesized briefly as follows: N-alpha-(tert-butyloxycarbonyl)-N-delta,N-omega-di-(benzyloxycarbonyl)-L-arginine (Boc-L-Arg(Di-Z)-OH) was dissolved in ethyl acetate (∼4% w/w) followed by addition of triphosgene (moles triphosgene/moles of Boc-L-Arg(Di-Z)-OH = 0.35) under strong stirring and inert atmosphere at room temperature. After 10 minutes, an equivalent amount of triethylamine was added (in relation to Boc-L-Arg(Di-Z)-OH), the solution was immersed in an ice bath and was left for 5 hours. The unreacted species along with the HCl and the amino acid salts were removed by extraction with an aqueous alkali solution and water. The organic phase was introduced into a specially designed custom-made apparatus for extreme purification by three crystallizations with ethyl acetate and n-hexane under high vacuum conditions. The formed L-Arg(Di-Z)-NCA was stored at inert atmosphere at 0 °C.
The reactions used for the synthesis of the NCAs are given in Schemes 1 and 2.
 | ||
| Scheme 1 Reactions used for the synthesis of the N-delta,N-omega-di-(benzyloxycarbonyl)-L-arginine N-carboxy anhydride. | ||
 | ||
| Scheme 2 Reactions used for the synthesis of the L-alanine N-carboxy anhydride. | ||
Synthesis of A12R2 oligopeptide
A custom-made glass apparatus was used without ground joints in order to create and maintain the conditions necessary for the living polymerization of NCAs.18,20 The polymerization reactors were designed to have a volume at least three times larger than the volume of the CO2 generated by each polymerization. Polymerizations were carried out with dimethylamine as the initiator. The sequential addition methodology was used for the synthesis of the oligopeptide. In a solution of L-Arg(Di-Z)-NCA in DMF (∼10% w/w), the appropriate amount of dimethylamine was added. After the completion of polymerization (1 day), a small aliquot was removed for characterization of the formed dimer of L-Arg(Di-Z). L-Ala-NCA was then added and was left to react for two days until all monomer was completely consumed. After complete consumption of Ala-NCA (2 days), a small aliquot of the raw product was removed from the reactor for characterization and control of the completion of the polymerization. The final oligopeptide was precipitated in diethylether, filtered and dried to constant weight.The deprotection of the benzyloxycarbonyl groups of arginine was performed by dissolving the oligopeptide in trifluoroacetic acid followed by addition of a solution of HBr in acetic acid (10 equivalents for each equivalent of protection). The solution was stirred at 0 °C for one hour. The oligopeptide was precipitated in diethyl ether, and was centrifuged to be collected quantitatively, and the solid was rinsed three times with diethyl ether. The oligopeptide was dried, and then was suspended in water, was placed in a dialysis bag and was dialyzed against MilliQ water six times. The solution was then lyophilized to give the oligopeptide (70% yield). The reactions used are given in Scheme 3.
 | ||
| Scheme 3 Reactions used for the synthesis of the A12R2 oligopeptide. | ||
Characterization of product
The protected oligopeptides were characterized by SEC and NMR spectroscopy. A special set of columns for low molecular weight samples were used to characterize the oligomers. The SEC system was composed of a Waters 600 high pressure liquid chromatographic pump, Waters HT columns, a Waters 410 differential refractometer detector and a Precision PD 2020 two angles (15°, 90°) light scattering detector. A 0.1 N LiBr DMF solution was used as an eluent at a rate of 1 mL min−1 at 60 °C. The SEC chromatograms obtained by the R2 precursor along with the final A12R2 are shown in ESI Fig. S1.† The molecular weight of the R2 precursor obtained was 930 Da and the one of the oligopeptide was 1760 Da, close to the one expected from stoichiometry (1745 Da).The composition of the oligopeptide was verified by 1H NMR spectroscopy. The peaks used to obtain the composition were at 5.11 ppm which is the aliphatic –CH2 protons of the benzyl groups of the protected L-arginine and the one at 4.1 ppm which is the total α-protons of the amino acid groups. The efficiency of the deprotection of L-arginine was monitored with FT-IR and 1H NMR spectroscopy. The deprotection of the L-arginine was verified from the elimination of the peaks at 5.11 ppm (aliphatic –CH2 protons of benzyl group) along with the peaks at 7.3 ppm which corresponds to the aromatic protons of the protective groups. In the FT-IR spectrum, the significant reduction of the peaks at 698 and 738 cm−1 indicate the deprotection of the L-arginine.
pH values
To characterize self-assembly in water, solutions were made using amounts of peptide dissolved in ultrafiltered (18 MΩ) water from a Barnstead Nanopure system. The pH values for solutions containing 0.5, 1 and 2 wt% A12R2 were measured (Mettler Toledo FiveEasy pH Meter) to be pH 4.12, pH 3.38 and pH 2.86 respectively.FTIR
Spectra were recorded using a Nexus FTIR spectrometer equipped with a DTGS detector and a multiple reflection attenuated total reflectance (ATR) system. Solutions of the peptide in D2O (1–2.5 wt%) were sandwiched in ring spacers between two CaF2 plate windows (spacer 0.006 mm). A spectrum was also measured for a film dried from a 0.5 wt% solution onto a CaF2 plate. All spectra were scanned 128 times over the range of 4000–950 cm−1.Circular dichroism
Spectra were recorded using a Chirascan spectropolarimeter (Applied Photophysics, UK). CD was performed on solutions of A12R2 in water which were placed in cover slip cuvettes (0.01 mm thick). Spectra are presented with absorbance A < 2 at any measured point with a 0.5 nm step, 1 nm bandwidth and 1 second collection time per step at 20 °C, taking four averages.X-ray diffraction (XRD)
Measurements were performed on stalks prepared by drying filaments of the peptide from 2 wt% solutions. Solutions of the peptide were suspended between the ends of wax-coated capillaries and dried. The stalks were mounted (vertically, with a 90 mm sample–detector distance) onto the four axis goniometer of a RAXIS IV++ X-ray diffractometer (Rigaku) equipped with a rotating anode generator. The XRD data was collected using a Saturn 992 CCD camera. The X-ray wavelength was λ = 1.54 Å. The same setup was used to measure XRD from a hydrated gel containing 17.4 wt% A12R2 which was prodded into a borosilicate capillary with D = 1 mm internal diameter and 0.01 mm wall thickness. The sample-to-detector distance was 50 mm.Cryo-transmission electron microscopy (cryo-TEM)
Cryo-transmission electron microscopy (cryo-TEM) was carried out using a field emission cryo-electron microscope (JEOL JEM-3200FSC), operating at 200 kV voltage. Images were taken in bright field mode and using zero loss energy filtering (omega type) with a slit width 20 eV. Micrographs were recorded using a Gatan Ultrascan 4000 CCD camera. The specimen temperature was maintained at −187 °C during the imaging. Vitrified specimens were prepared using an automated FEI Vitrobot device using Quantifoil 3.5/1 holey carbon copper grids with a hole size of 3.5 μm. Just prior to use, grids were plasma cleaned using a Gatan Solarus 9500 plasma cleaner and then transferred into an environmental chamber of a FEI Vitrobot having room temperature and 100% humidity. Thereafter 3 μL of sample solution (2 wt% concentration) was applied on the grid and it was blotted twice for 5 seconds and then vitrified in a 1/1 mixture of liquid ethane and propane at temperature of −180 °C. The most viscous gel (5 wt% concentration) was blotted 4 times for 5 s. The grids with vitrified sample solution were maintained at liquid nitrogen temperature and then cryo transferred in to the microscope.Small-angle and wide-angle X-ray scattering
SAXS/WAXS data were collected on beamline ID02 at the ESRF, Grenoble, France. SAXS data were collected with a FReLoN Kodak CCD with a 1.2 m sample–detector distance and WAXS data were measured simultaneously with an Aviex CCD. The X-ray wavelength was 0.995 Å. More dilute solution samples (0.5 wt%, 1 wt% and 2 wt%) were injected using a syringe into ENKI KI-beam thin (0.05 mm) wall 1.85 mm diameter polycarbonate capillaries which optimise background subtraction. Measurements were performed at 25 °C. Gels of 5 wt% and 10 wt% samples were placed within a PTFE-spacer ring between PET films and mounted into sealed aluminium cells. Measurements were performed at 20 °C and 40 °C. Additional data (not shown) were collected on the bioSAXS beamline BM29 at the ESRF, Grenoble, France. Solutions containing 0.5 wt% or 1 wt% A12R2 were loaded in PCR tubes in an automated sample changer. SAXS data was collected using a Pilatus 1M detector. The sample–detector distance was 2.84 m. The X-ray wavelength was 0.99 Å. All data were reduced to one-dimensional intensity profiles by radial integration.Results
The oligomer A12R2 was successfully synthesized using NCA polymerization methods, as summarized in Scheme 1. Bulk protective groups such as two benzyl groups were intentionally chosen for the protection of the arginine residues in order to have low propagation rate of the polymerization of the corresponding NCA compared to the initiation rate, and thus form mainly the dimer.Spectroscopic methods (CD and FTIR) were used to investigate secondary structure formation. FTIR spectra in the amide I′ region are shown in Fig. 1 and reveal a strong peak in the spectra for all three samples (0.5 wt%, 1 wt% and 2 wt%) at 1626 cm−1, which is ascribed to predominant β-sheet ordering.21,22 The peak at 1652 cm−1 may be assigned to a minority of α-helical structure (vide infra, there is also evidence from XRD for α-helical content).21,22 The shoulder peak at 1693–1694 cm−1 may be assigned to antiparallel β-sheet structure.21,23–25 There is also a peak in the amide II′ band at 1545 cm−1. The amide I′ region features are distinct from those for A6K, which only shows β-sheet features at high concentration, above about 15 wt%, above the critical aggregation concentration.10,11 Interestingly, the peak at 1608 cm−1 associated with the arginine side chain24,25 is not observed pointing to salt binding interactions.
 | ||
| Fig. 1 FTIR spectra for A12R2 in aqueous solution at the concentrations shown. | ||
CD spectra are also consistent with β-sheet structure, which increases with concentration. The spectra shown in Fig. 2 show a maximum at 195 nm and a minimum at 218 nm, these features being diagnostic of significant β-sheet content.26–28
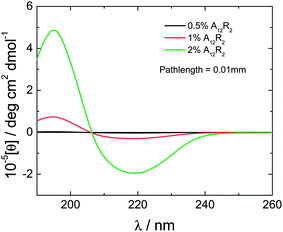 | ||
| Fig. 2 CD spectra for A12R2 in aqueous solution at the concentrations indicated. | ||
Cryo-TEM revealed a mesh of fine fibrils covering the vitreous layer as shown in Fig. 3 which includes a representative image for a 2 wt% sample. In some regions where the film is thinner, areas showing single layers of fibrils could be observed (ESI Fig. S2,† includes typical images for a 5 wt% gel). In all cases, the diameter of the fibrils could be estimated at 5–6 nm. At high magnification, the fibrils appear to be twisted with a pitch of ca. 10 nm. The dense mesh of arginine-functionalized ultrathin fibrils obtained by self-assembly of A12R2 may be useful in applications such as antimicrobial coatings.
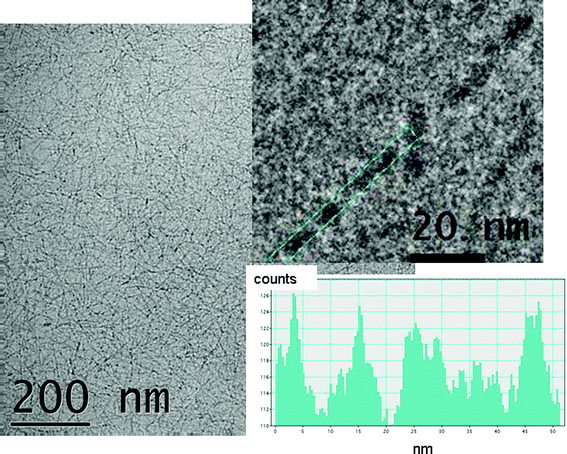 | ||
| Fig. 3 Cryo-TEM image from a 2 wt% solution of A12R2. The top inset shows an enlargement showing a helical twist with a pitch of (12 ± 2) nm and the bottom inset shows the corresponding intensity profile. | ||
SAXS intensity (I) profiles are shown in Fig. 4. The shape of the profiles was similar for all samples investigated in the concentration range 0.5–10 wt%, indicating a similar self-assembled structure is formed. Actually, the profiles measured under the same conditions, i.e. the dilute samples in capillaries superpose when adjusted for concentration in an I/c plot. The intensity profiles are consistent with a fibrillar structure as shown by the fit using a cylinder form factor. The fit parameters were cylinder radius R = 2.5 nm (length L = 25 nm fixed, kept relatively short consistent with the TEM images) with Gaussian polydispersity ΔR = 40%, relative electron density contrast η = 0.0001 and constant background BG = 0.0001. The radius agrees very well with the fibril diameter from cryo-TEM.
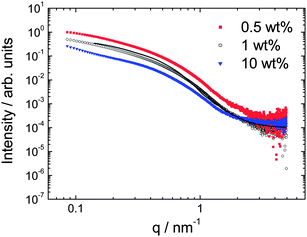 | ||
| Fig. 4 SAXS data at the concentrations indicated (measured at 20 °C for 0.5 wt% and 1 wt% and 25 °C for 10 wt%). The data for 0.5 and 1 wt% samples is for solutions mounted in capillaries, that for 10 wt% is for a gel mounted in a sandwich cell, the high q region of this data is influenced by the background subtraction. The line through the data for the 1 wt% sample is a fit to a cylinder form factor, as described in the text. | ||
Fibre X-ray diffraction was performed to investigate the self-assembled morphology at the level of the β-sheet structure. For the dried stalk, alignment was observed in the X-ray pattern as shown in Fig. 5(a). There are equatorial reflections corresponding to d = 7.7 A and 5.4 Å, and a strong meridional reflection at d = 4.42 Å. A further set of four off-axis reflections is observed at d = 3.74 Å. To quantify peak positions, one-dimensional integration was performed, the corresponding profiles being shown in Fig. 5(b) along with profiles for the hydrogel which did not show alignment in the 2D pattern. The hydrogel shows the same series of reflections as observed for the dried stalk, confirming that drying does not introduce artifacts. This was also confirmed by in situ synchrotron SAXS (ESI Fig. S3†) which shows reflections 7.6, 5.3 and 4.3 Å. Reflections at 5.5, 4.5 and 3.8 Å were reported for the related peptide A6K.10 However that peptide forms thin wall nanotubes in contrast to A12R2. However, the 5.3–5.5 Å reflection for the two peptides is assigned to a common feature, the stacking distance of β-sheets and the 4.3–4.4 Å reflection is due to the β-strand spacing. The 5.3–5.5 Å spacing is consistent with the 5.3 Å reflection observed for alanine-rich peptides29 and the polyalanine crystal structure.30,31 The spacing is much lower than the typical β-sheet stacking distance due to the efficient packing of small alanine residues. The spacing of the 4.4 Å reflection for A12R2 is also smaller than the usual 4.7–4.8 Å spacing of β-strands,32 and is consistent with a more tightly packed structure as observed for other alanine-rich peptides,29 and indicates shorter hydrogen bonds. The 7.7 Å repeat period (Fig. 6) has previously been assigned to the (100) reflection from an α-helical structure, coexisting with β-sheet structure for oligomeric alanine peptides (9–19 repeats)31 and the same assignment is used here. The 3.74 Å reflections appear not to be second order reflections from the 7.7 Å peak, first due to the difference in spacing with respect to 7.7 Å/2 but mainly due to the off-axis location of the reflections (not observed for the 7.7 Å reflection). Instead, they are ascribed to diffraction from planes containing Cα moieties.32,33 Our assignments of the observed periods are consistent with the orientation of the reflections shown in Fig. 5(b),34i.e. the 7.7 and 5.4 Å periodicities are perpendicular to the fibril axis, whereas the 4.4 Å (H-bonding) spacing is parallel to the fibril axis.32,33Fig. 6 shows a simplified schematic for the proposed structure that accounts for the 5 nm fibril width observed by both cryo-TEM and SAXS and the spacings (and orientation of reflections) obtained from XRD. In Fig. 6, only two stacked twisted tapes within the fibrils are shown, in reality there will be more (at least ∼10 based on the corresponding Bragg peak width) in order that a 5.4 Å Bragg reflection is observed, and to produce a 5 nm diameter fibril. The propeller-like twisting within the fibrils results from the electrostatic repulsion between the terminal R2 units within the antiparallel A12R2 dimers that are proposed as the fundamental structural building block (Fig. 6). These are stabilized by strong hydrogen bonding of alanine residues and efficient stacking of the small methyl side chains, as well as the electrostatic repulsion of the arginine units which is minimized with an antiparallel configuration. The observation of fibrils based on stacked ribbons is consistent with prior reports,35 however the basic stacking unit based on dimer-wide tapes is unprecedented to our knowledge.
 | ||
| Fig. 5 (a) Fibre X-ray diffraction pattern measured from a stalk dried from a 2 wt% solution. (b) Fibre X-ray diffraction intensity profiles measured with a dried stalk, or from a 17.4 wt% in situ hydrogel. | ||
 | ||
| Fig. 6 Proposed model showing ribbon (left) with width equal to an antiparallel A12R2 dimer (right). The A2 units are indicated in the ribbon schematic in red. The β-strand spacing is 4.4 Å (not to scale), and the pitch of the helix is ca. 10 nm. The β-sheet stacking distance (displacement between tapes) is 5.3 Å, only two stacked tapes are shown for clarity. | ||
Summary and conclusions
Distinctly from nanotube-forming peptides A6K10 and A6R,36 the surfactant-like peptide A12R2 forms fibrils in aqueous solution at low concentration. These fibrils have an unusually small diameter, 5–6 nm, compared to typical amyloid fibrils. FTIR shows that A12R2 forms antiparallel β-sheets. XRD provides the spacings of the cross-β structure. The strand spacing, 4.4 Å is smaller than that typically observed, as is the β-sheet stacking distance. The presence of a (minority, based on FTIR spectra) fraction of α-helical content leads to a 7.7 Å spacing, corresponding to the lateral spacing of the helices.The self-assembly of the peptide A12R2 is governed by a balance between the desire to reduce the electrostatic repulsion of the terminal arginine residues which produces an antiparallel dimer, and also leads to a twist of the fibrils and the stacking of the alanine residues. The repeating alanine sequence leads to closely stacked β-strands and β-sheets due to the efficient steric packing of the methyl side chains. These findings are in agreement with reports on alanine-rich silk-mimic peptides29 and polyalanine,30,31 and point towards the possibly generality of this mode of packing in peptides with multiple alanine repeats. In contrast to these prior reports the A12R2 peptide has been designed to be amphiphilic, incorporating a cationic di-arginine headgroup.
The structure detailed in this work provides the basis to rationally design other ultra-thin amyloid peptide fibrils. The arginine coating of the A12R2 fibrils is presented at high density and this has potential for functionalization in a variety of applications. For example the bidentate nature of the arginine guanidinium units leads to favorable interactions with phosphate groups. We also note that the fibrils form a high density “mat-like” structure at high oligopeptide concentration, suggesting possible applications in antimicrobial coatings since antimicrobial peptides are generally arginine-rich. These and related bionanoscience applications are under ongoing investigation in our group.
Acknowledgements
The synthesis of the peptide was supported by the European Soft Matter Infrastructure Initiative (ESMI, proposal reference S110700102). Work in the Hamley lab was supported by EPSRC grants EP/G026203/1 and EP/G067538/1. Beamtime at the ESRF on beamline ID02 was awarded under reference SC3468 and on BM29 under reference MX-1401. We are grateful to Nick Spencer for assistance with XRD experiments, Theyencheri Narayanan for assistance on ID02 and Petra Pernot on BM29. We thank Dr Franck Artzner (Université de Rennes, France) for helpful comments.References
- S. Vauthey, S. Santoso, H. Gong, N. Watson and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5355–5360 CrossRef CAS.
- S. Santoso, W. Hwang, H. Hartman and S. Zhang, Nano Lett., 2002, 2, 687–691 CrossRef CAS.
- X. B. Zhao, F. Pan, H. Xu, M. Yaseen, H. H. Shan, C. A. E. Hauser, S. G. Zhang and J. R. Lu, Chem. Soc. Rev., 2010, 39, 3480–3498 RSC.
- I. W. Hamley, Soft Matter, 2011, 7, 4122–4138 RSC.
- S. G. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS.
- X. J. Zhao, Y. Nagai, P. J. Reeves, P. Kiley, H. G. Khorana and S. G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17707–17712 CrossRef CAS.
- X. Q. Wang, K. Corin, P. Baaske, C. J. Wienken, M. Jerabek-Willemsen, S. Duhr, D. Braun and S. G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9049–9054 CrossRef CAS.
- G. von Maltzahn, S. Vauthey, S. Santoso and S. Zhang, Langmuir, 2003, 19, 4332–4337 CrossRef CAS.
- A. Nagai, Y. Nagai, H. J. Qu and S. G. Zhang, J. Nanosci. Nanotechnol., 2007, 7, 2246–2252 CrossRef CAS.
- V. Castelletto, D. R. Nutt, I. W. Hamley, S. Bucak, C. Cenker and U. Olsson, Chem. Commun., 2010, 46, 6270–6272 RSC.
- S. Bucak, C. Cenker, I. Nasir, U. Olsson and M. Zackrisson, Langmuir, 2009, 25, 4262–4265 CrossRef CAS.
- R. L. Thurlkill, G. R. Grimsley, J. M. Scholtz and C. N. Pace, Protein Sci., 2006, 15, 1214–1218 CrossRef CAS.
- K. Zhao, U. J. Choe, D. T. Kamei and G. C. L. Wong, Soft Matter, 2012, 8, 6430–6433 RSC.
- J. S. Wadia, R. V. Stan and S. F. Dowdy, Nat. Med., 2004, 10, 310–315 CrossRef CAS.
- D. Derossi, G. Chassaing and A. Prochiantz, Trends Cell Biol., 1998, 8, 84–87 CAS.
- N. Schmidt, A. Mishra, G. H. Lai and G. C. L. Wong, FEBS Lett., 2009, 584, 1806–1813 CrossRef.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, S. Pispas and M. Pitsikalis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3211–3234 CrossRef CAS.
- A. Gitsas, G. Floudas, M. Mondeshki, H. W. Spiess, T. Aliferis, H. Iatrou and N. Hadjichristidis, Macromolecules, 2008, 41, 8072–8080 CrossRef CAS.
- T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2004, 5, 1653–1656 CrossRef CAS.
- P. Haris and D. Chapman, Biopolymers, 1995, 37, 251–263 CrossRef CAS.
- B. Stuart, Biological Applications of Infrared Spectroscopy, Wiley, Chichester, 1997 Search PubMed.
- S. Krimm and J. Bandekar, Adv. Protein. Chem., 1986, 38, 181–364 CrossRef CAS.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS.
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369–430 CrossRef CAS.
- R. W. Woody, Methods Enzymol., 1995, 246, 34–71 CAS.
- B. M. Bulheller, A. Rodger and J. D. Hirst, Phys. Chem. Chem. Phys., 2007, 9, 2020–2035 RSC.
- B. Nordén, A. Rodger and T. R. Dafforn, Linear Dichroism and Circular Dichroism: A Textbook on Polarized-Light Spectroscopy, RSC, Cambridge, 2010 Search PubMed.
- O. Rathore and D. Y. Sogah, J. Am. Chem. Soc., 2001, 123, 5231–5239 CrossRef CAS.
- S. Arnott, S. D. Dover and A. Elliott, J. Mol. Biol., 1967, 30, 201–208 CrossRef CAS.
- A. Fujie, T. Komoto, M. Oya and T. Kawai, Makromol. Chem., 1973, 169, 301–321 CrossRef CAS.
- M. Sunde, L. C. Serpell, M. Bartlam, P. E. Fraser, M. B. Pepys and C. C. F. Blake, J. Mol. Biol., 1997, 273, 729–739 CrossRef CAS.
- L. C. Serpell, Biochim. Biophys. Acta, Bioenerg., 2000, 1502, 16–30 CrossRef CAS.
- T. E. Creighton, Proteins: Structures and Molecular Properties, W.H. Freeman, New York, 1993 Search PubMed.
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. B. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857–11862 CrossRef CAS.
- I. W. Hamley, A. Dehsorkhi and V. Castelletto, Chem. Commun., 2013, 49, 1850–1852 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sm50303h |
| This journal is © The Royal Society of Chemistry 2013 |