 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low friction and high load bearing capacity layers formed by cationic-block-non-ionic bottle-brush copolymers in aqueous media†
Xiaoyan
Liu
*a,
Esben
Thormann
a,
Andra
Dedinaite
ab,
Mark
Rutland
ab,
Ceslav
Visnevskij
c,
Ricardas
Makuska
c and
Per M.
Claesson
ab
aKTH Royal Institute of Technology, School of Chemical Sciences and Engineering, Department of Chemistry, Surface and Corrosion Science, Drottning Kristinas väg 51, SE-100 44 Stockholm, Sweden. E-mail: xiaoyanl@kth.se
bSP Technical Research Institute of Sweden, Chemistry, Materials and Surfaces, P.O. Box 5607, SE-114 86 Stockholm, Sweden
cDepartment of Polymer Chemistry, Vilnius University, Naugarduko 24, LT-03225 Vilnius, Lithuania
First published on 17th April 2013
Abstract
Efficient lubricants should be able to build surface layers that result in low friction and high load bearing capacity. In this work we show how this can be achieved in aqueous media by means of adsorption of a diblock copolymer consisting of a cationic anchor block without side chains and an uncharged and hydrophilic bottle-brush block that protrudes into solution. Surface and friction forces were measured between negatively charged silica surfaces coated with adsorbed layers of the cationic diblock copolymer, utilizing the atomic force microscope colloidal probe technique. The interactions between the surfaces coated with this copolymer in water are purely repulsive, due to a combination of steric and electrostatic double-layer forces, and no hysteresis is observed between forces measured on approach and separation. Friction forces between the diblock copolymer layers are characterized by a low friction coefficient, μ ≈ 0.03–0.04. The layers remain intact under high load and shear due to the strong electrostatic anchoring, and no destruction of the layer was noted even under the highest pressure employed (about 50 MPa). Addition of NaCl to a concentration of 155 mM weakens the anchoring of the copolymer to the substrate surface, and as a result the friction force increases.
1 Introduction
Nature has developed efficient aqueous lubrication systems based on phospholipids combined with biopolymers.1–3 Notably, bottle-brush structures in molecules such as lubricin3,4 and hyaluronan–aggrecan association structures play important roles in, for example, joint lubrication.5–7 These findings have inspired the synthesis of polymers with similar architectures, and also some of these have been shown to provide favorable lubrication properties.8–10Dense brush layers that experience good solvency conditions are another alternative to achieve low friction forces.9–11 For instance, recently, polyzwitterionic brush layers were shown to provide a very low coefficient of friction (μ ≈ 0.0004) up to a mean pressure of 7.5 MPa.12 High-density brush layers are most easily prepared by grafting methods, and particularly using grafting-from rather than grafting-to methods.10 However, this surface preparation method is relatively complicated and the layers have no self-healing capacity. Extended polymer layers with good lubrication properties can also be obtained by adsorption from solution using bottle-brush polymers.4,10,13 Such layers will normally have a lower chain density than layers obtained by grafting methods, but the simple layer formation process and the self-healing capacity of self-assembled layers are two advantages with this approach.
Poly(ethylene oxide) (PEO), also known as poly(ethylene glycol) (PEG), is one of the most studied water-soluble polymers in terms of both aqueous solution14,15 and interfacial properties.16–18 PEO plays a central role in many applications due to its efficacy in stabilizing colloidal particles.19 Recently, the random copolymer poly(L-lysine)-g-poly(ethylene glycol), PLL-g-PEG, has been shown to provide low friction layers on silicon oxide surfaces where the PEG chains are forced into a dense, brush-like structure.20 Systematic studies of bottle-brush polyelectrolytes with PEO side chains attached randomly along a cationic backbone have also been presented by our group. Bottle-brush polyelectrolytes with PEO side chains randomly attached to the main chain have successfully been used for achieving low protein adsorption,21–23 strong steric repulsion,4 and low friction forces.4,24
Modelling has suggested that the adsorbed layer structure differs significantly depending on whether the PEO side chains are randomly distributed along the backbone, or assembled at one end of the polymer chain forming a diblock copolymer structure. In particular, compared to the case with a random distribution of PEO side chains along the cationic backbone that has been exclusively studied in lubrication applications so far (Fig. 1c), a more highly extended and water-rich layer is expected for a block copolymer structure consisting of a cationic block without any side chains and an uncharged block with a high density of PEO side chains (Fig. 1b).25,26 Inspired by the theoretical predictions we have designed a diblock copolymer consisting of a cationic block without any side chains and an uncharged bottle-brush block where each main-chain segment carries a 45 unit long PEO side chain. The synthesis and adsorption properties of this block copolymer were recently presented, and it was indeed shown that a significantly more extended adsorbed layer was formed by this polymer, compared to that formed by random copolymers built from the same two segment types.27 The layer formed by the diblock copolymer was described as a branched brush layer in recognition of the preferential adsorption of the cationic block to the surface, presenting a branched bottle-brush structure towards the solution.
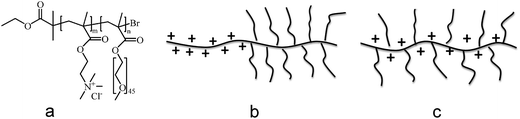 | ||
| Fig. 1 (a) Molecular structure of (METAC)m-b-(PEO45MEMA)n, (b) sketch of the structure of (METAC)m-b-(PEO45MEMA)n, (c) sketch of an example of a random copolymer with PEO side chains along the cationic backbone. | ||
In this paper, we report the lubricating ability of these branched brush layers. Using the AFM colloidal probe technique we evaluate (i) the friction coefficient, (ii) the load bearing capacity, and (iii) structural changes within the adsorbed layer due to the combined action of load and shear, and how addition of an electrolyte that reduces the polymer–surface affinity affects these properties.
2 Materials and methods
2.1 Materials
The diblock copolymer [(METAC)m-b-(PEO45MEMA)n] (Fig. 1a) was synthesized by AGET ATRP (activators generated by electron transfer, atom transfer radical polymerization) as described elsewhere.27 In this representation, METAC stands for methacryloxyethyl trimethylammonium chloride and PEO45MEMA stands for poly(ethyleneoxide)45 methylether methacrylate. The number average degree of polymerization of the METAC block is 90 (polydispersity index 1.25), whereas that of the non-ionic block is 100, with a high polydispersity. The number average polymer molecular weight is 230 kDa.Sodium chloride (NaCl, BioXtra, ≥99.5%) was purchased from Sigma-Aldrich. Water was purified by employing a Milli-ROPls unit connected to a Milli-Q plus 185 system and filtered through a 0.2 μm Millipak filter at 25 °C. The resistivity of the water was 18.2 MΩ cm and the organic content was less than 3 ppb.
Thermally oxidized silicon wafers with a 100 nm thick SiO2 layer from Wafer Net, Germany, were used as the flat substrate in AFM measurements. The wafers were cut to size and cleaned by immersion in 2% Hellmanex (Hellma GmbH) solution for 30 minutes followed by rinsing several times with Milli-Q water. The wafers were left overnight in Milli-Q water before measurements.
Silica particles of approximately 20 μm in diameter from Duke Scientific Corp. were employed as colloidal probes. The diameter of the probe was determined by optical microscopy. The cantilevers with silica particles were cleaned in a plasma cleaner (Harrick PDC-3XG, New York) for 30 seconds.
2.2 Methods
The NanoScope Analysis Version 1.20 (Bruker) software was employed to analyze the recorded images and extract topographical and mechanical properties. The height images were flattened to remove tilt prior to image analysis. The surfaces were imaged using a scan size of 500 nm. In each case, a peak force of 1 nN and a scan rate of 0.5 Hz were used. The PeakForce QNM experiments were carried out in 4 consecutive steps, following the same sequence as for the force and friction measurements. First, the silica surface was imaged in water. Next, a 50 ppm solution of (METAC)m-b-(PEO45MEMA)n in water was injected and the diblock copolymer was allowed to adsorb for two hours. The diblock copolymer layer on the silica surface was then imaged first with polymer present in solution, and then after replacing the polymer solution with water. It was finally also imaged in 155 mM NaCl solution.
Before each experiment, the fused silica liquid cell and all other tools were cleaned by immersion in 2% Hellmanex (Hellma GmbH) solution for about 1 hour. They were then rinsed several times with Milli-Q water and blow-dried with filtered nitrogen gas. The AFM experiments were started by measuring surface forces between the silica surface and the silica probe in water. After this, layers of (METAC)m-b-(PEO45MEMA)n were allowed to form on the silica surface and the silica probe by adsorption from a 50 ppm solution of (METAC)m-b-(PEO45MEMA)n in water for 2 hours. Next, the surface forces were measured, and the system was left to equilibrate for 10 minutes, after which the surface forces were measured again, followed by friction measurements (repeated twice with the same settings directly after each other). Surface forces were measured again following the friction experiments. Next, the layers were rinsed with Milli-Q water and the experiment followed the same procedure as described for the case with diblock copolymer solution in the AFM liquid cell. Finally, 155 mM NaCl solution was employed to rinse the system and the same experimental procedure was carried out again.
The surface forces were measured with a constant approach and retraction speed of 2 μm s−1. The reproducibility of the surface force data is illustrated in the ESI (Fig. S1†). The friction measurements were performed by sliding the surfaces backwards and forwards ten times at each normal load and registering the cantilever twist angle. The sliding distance was 1 μm in each direction and the scan rate was 1 Hz, giving a sliding speed of 2 μm s−1.
3 Results
Before describing the results of this study, we recapitulate some relevant data (see Table 1) for the adsorbed (METAC)m-b-(PEO45MEMA)n layer obtained from QCM-D and optical reflectometry measurements as reported in our previous work.27Rinsing with water hardly affects the structure of the adsorbed layer as judged from the adsorbed amount, layer thickness and water content. However, rinsing with 100 mM NaCl leads to minor desorption and a small decrease in layer thickness and water content. This is due to decreased electrostatic affinity to the surface. We note the very high water content, >90%, of the undisturbed polymer layer. The water content is expected to decrease with applied load, but this cannot be quantified with the technique used here. However, it could be assessed by means of a surface force apparatus (SFA), for example, and such studies could shed further light on the importance of hydration water for boundary lubrication by means of adsorbed polymer layers.
3.1 PeakForce QNM imaging
The topography and local variations in deformation of the adsorbed layer of (METAC)m-b-(PEO45MEMA)n on silica were probed by PeakForce QNM. The topographical images and the corresponding deformation images for bare silica in water and for the adsorbed polymer layer are shown in Fig. 2. It is clearly observed that both topography and deformation change when the diblock copolymer layer is formed (see Fig. 2b and d). | ||
| Fig. 2 PeakForce QNM mode topographical images of (a) silica surface in water, (b) diblock copolymer (METAC)m-b-(PEO45MEMA)n adsorbed on the silica surface. The deformation images of (c) silica surface in water and (d) the diblock copolymer adsorbed on the silica surface. The scale bar of 200 nm is identical for all images. | ||
Height and deformation information extracted from such images are summarized in Table 2. We note the smoothness of the silica substrate where the peak-to-valley distance is about 1 nm, and the Rq value is below 0.2 nm. The hard nature of the silica surface results in low deformation, 1.8 nm, and only very small variation across the substrate (the Rq value of the deformation image is 0.04 nm). Adsorption of the diblock copolymer results in a somewhat increased roughness, Rq = 0.4 nm, and a significant increase in deformation to 4.8 nm. The deformation decreases slightly when the layer is rinsed with water and further upon rinsing with 155 mM NaCl. The tip–surface adhesion and the energy dissipation are in all cases close to zero and therefore not reported in Table 2.
| Surface | Height (nm) | Deformation (nm) | |
|---|---|---|---|
| Silica in water | Mean value | 0 | 1.8 |
| R q | 0.18 | 0.04 | |
| Adsorbed layer with polymer present in solution | Mean value | 0 | 4.8 |
| R q | 0.38 | 0.6 | |
| Adsorbed layer after rinsing with water | Mean value | 0 | 4.1 |
| R q | 0.4 | 0.72 | |
| Adsorbed layer after rinsing with 155 mM NaCl | Mean value | 0 | 3.2 |
| R q | 0.37 | 0.67 | |
3.2 Surface and friction forces
 | ||
| Fig. 3 Force normalized by radius as a function of separation between two silica surfaces across water. Filled and unfilled symbols represent data obtained on approach and retraction, respectively. The upper and lower lines are fitted DLVO forces using constant charge and constant potential boundary conditions, respectively. The fitted Debye-length of 107 nm corresponds to a salt concentration of 8 × 10−6 M. | ||
The forces measured between the silica probe and the silica surface in the presence of the diblock copolymer in solution are shown in Fig. 4, together with calculated DLVO force curves. The most long range part of the force curve is consistent with a double-layer force. However, at separations below 40 nm, the force increases more steeply, demonstrating the presence of a long range steric force. The apparent double-layer potential was found to have a magnitude of 33 mV at the onset of the steric interaction at a separation of 40 nm. There is no detectable sign of hysteresis between forces measured on approach and retraction.
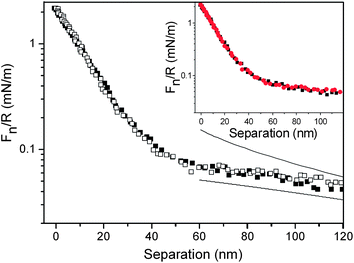 | ||
| Fig. 4 Force normalized by radius as a function of separation between silica surfaces coated with (METAC)m-b-(PEO45MEMA)n in the presence of 50 ppm of the diblock copolymer in solution. Filled and unfilled symbols represent data points obtained on approach and retraction, respectively. The upper and lower lines are fitted DLVO forces using constant charge and constant potential boundary conditions, respectively. The fitted Debye-length of 107 nm corresponds to a salt concentration of 8 × 10−6 M. The inset shows the forces acting between the polymer layers before (black squares) and after (red circles) rinsing with water. In this figure, zero distance is set to the separation between the surfaces under the highest normalized force used in the experiment (about 2 mN m−1). | ||
The inset in Fig. 4 provides a comparison of the forces measured on approach in the 50 ppm diblock copolymer solution with those determined after rinsing away the polymer from bulk solution. The limited effect of rinsing away the diblock copolymer from bulk solution is consistent with the very limited desorption noted by optical reflectometry (see Table 1).
The practically irreversible nature of the adsorption allows us to preadsorb the diblock copolymer layer on the flat silica surface, and then investigate the interaction between this layer and a bare silica colloidal probe. From such force measurements it is possible to determine the sign of the double-layer potential of the polymer-coated surface. The measured forces in this asymmetric system are shown in Fig. 5, where the inset also shows forces calculated by using the DLVO theory. The long range repulsion encountered on approach is consistent with a double-layer force where the surface interaction is described by the constant surface charge boundary condition, whereas the long-range attraction predicted by the constant surface potential model is not observed. In the calculated DLVO force curves, the silica surface potential was fixed at −70 mV (as determined from the data in Fig. 3). The offset value was set to 20 nm (which corresponds to the onset of bridging attraction, see below), and the Debye-length was set to 107 nm (identical to that found between two bare silica surfaces and between two diblock copolymer coated silica surfaces (see Fig. 3 and 4)). With this choice of calculation parameters the apparent double-layer potential at the diblock copolymer coated surface was found to be −23 mV. The magnitude of the potential would change if the offset value was varied, but the sign of the potential will always be negative. The fact that the surface potential of the diblock copolymer coated silica surface is negative demonstrates that excluded volume repulsion, and not electrostatic forces, limits the adsorption of (METAC)m-b-(PEO45MEMA)n on silica in pure water.
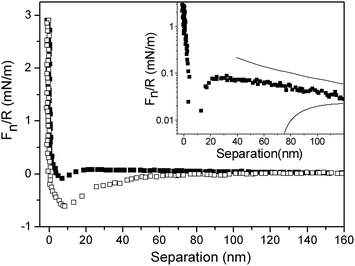 | ||
| Fig. 5 Force normalized by radius as a function of separation between a bare silica probe and a silica surface coated with (METAC)m-b-(PEO45MEMA)n in water. The filled and unfilled symbols correspond to forces measured on approach and separation, respectively. The inset shows the approach force curve and the corresponding forces calculated within the DLVO framework using constant charge (upper line) and constant potential (lower line) boundary conditions, respectively. | ||
The measured force reaches a maximum at a separation of 20 nm, and the minimum force is observed at a separation of 9 nm. We interpret these features as being due to a bridging attraction, where PEO chains from the diblock copolymer coated surface adsorb to the bare silica substrate. In contrast to the case with two polymer-coated surfaces (Fig. 4), a significant hysteresis can be found between compression and decompression, and this is attributed to the bridging force. Our interpretation finds support from the long-range nature of the attraction observed on separation, which is a sign of polymer stretching.38–41
The friction forces measured between the bare silica probe and the bare silica surface are reported in Fig. 6. The coefficient of friction is found to be 0.25, which is somewhat larger than the value reported for the silica–silica system in water (μ = 0.11) by Grant and Tiberg,42 but lower than the value of μ = 0.42 reported by McNamee et al.43 The large variation in the friction coefficient for silica–silica across water reported in the literature is likely a consequence of the different cleaning procedures used that result in different surface roughness and silanol group content.
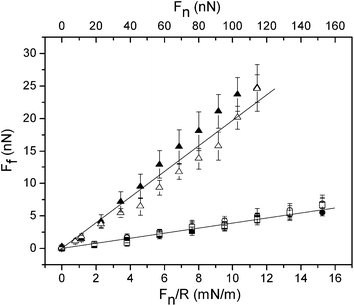 | ||
| Fig. 6 Friction force vs. load (Fn and Fn/R) curves observed for a silica probe and a silica surface in water (triangles) and after adsorption of (METAC)m-b-(PEO45MEMA)n from an aqueous 50 ppm solution. The first measurement cycle (squares) and the subsequent one (circles) are shown. Filled and unfilled symbols represent data obtained on loading and unloading, respectively. The straight lines are fits to the data points obtained at low loads. They are included for illustrating eventual deviations from Amontons' law. The error bars correspond to standard deviations from ten measurements. | ||
Two friction force vs. load curves measured between silica surfaces coated with (METAC)m-b-(PEO45MEMA)n in a 50 ppm solution of this diblock copolymer solution are illustrated in Fig. 6. The two curves are very similar, and the friction force at any explored load is significantly smaller than that between the bare silica surfaces. We suggest that the reduction of the friction force is attributed to the steric repulsion between the extended and water-rich branched brush layers formed by the diblock copolymer. The friction coefficient between the (METAC)m-b-(PEO45MEMA)n coated surfaces is much lower than that of the bare silica–silica system, about 0.04, and a close to Amontons-like behaviour, Ff = μFn, is observed. Each data point in Fig. 6, and in other figures displaying friction forces, is based on measurements of at least 10 consecutive friction loops. We note (i) the small error bars when the polymer is present on the surface, (ii) that very similar friction forces are observed in all friction loops, i.e. there is no run-in time for achieving low friction, and (iii) each friction loop is smooth providing evidence for the absence of significant stick–slip features.
The force curves measured between the silica surfaces coated with (METAC)m-b-(PEO45MEMA)n before and after friction measurements in the presence of this diblock copolymer in solution are presented in Fig. 7, together with force curves calculated by using the DLVO theory. There is no difference in the forces recorded before and after friction measurements, and in both cases no hysteresis between forces measured on approach and separation can be noticed. This demonstrates that the structure of the layers on the silica surfaces remains intact also under the combined action of load and shear, and they thus have a high load bearing capacity.
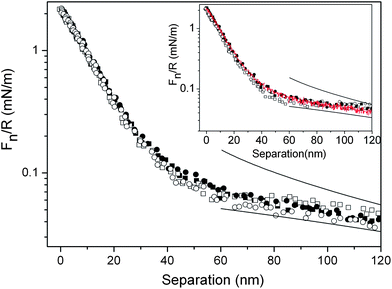 | ||
| Fig. 7 Force normalized by radius as a function of separation between silica surfaces coated with (METAC)m-b-(PEO45MEMA)n in the presence of 50 ppm of the diblock copolymer in solution. Squares denote data points obtained before friction measurements, and circles denote data obtained after friction measurements. Filled and unfilled symbols represent approach and retraction data, respectively. The upper and lower lines are fitted DLVO forces using constant charge and constant potential boundary conditions, respectively. The Debye-length was 107 nm, corresponding to a salt concentration of 8 × 10−6 M. The inset provides a comparison of the force curves after rinsing with water measured on approach (black filled symbols) and retraction (unfilled symbols) with those measured in the presence of the diblock copolymer in solution before friction measurements (red line). | ||
The force curves measured between silica surfaces coated with (METAC)m-b-(PEO45MEMA)n after rinsing away the polymer from bulk solution before and after friction measurements are shown in the inset of Fig. 7. There is still no difference in the surface forces encountered before and after friction measurements, and hardly any hysteresis between approach and retraction curves. This demonstrates that the layers formed on the surfaces are not permanently changed under the application of load and shearing, even when no polymer is present in bulk solution.
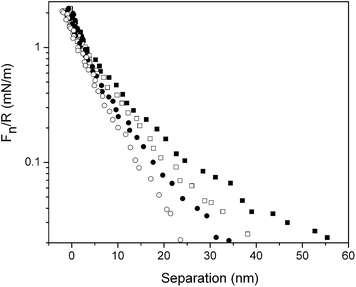 | ||
| Fig. 8 Force normalized by radius as a function of separation between silica surfaces coated with (METAC)m-b-(PEO45MEMA)n across a polymer-free 155 mM NaCl solution. Squares denote curves obtained before friction measurements, and circles denote curves obtained after friction measurements. Filled and unfilled symbols represent approach and retraction curves, respectively. | ||
Friction forces measured between silica surfaces coated with (METAC)m-b-(PEO45MEMA)n in water (no polymer in solution) and in 155 mM NaCl are compared in Fig. 9. The friction coefficient between the (METAC)m-b-(PEO45MEMA)n coated surfaces in water is about 0.03, and the results from surface forces and friction measurements show that the experiments have good reproducibility and the layers have a high load bearing capacity in water. The friction is significantly higher in 155 mM NaCl. We suggest that this is caused by energy dissipation due to breakage and reformation of cationic segment–surface attachment points, a process that is promoted by the decreased electrostatic surface affinity of the polymer layer due to the high ionic strength of the solution. Note, however, that similar friction forces are encountered on loading and unloading, suggesting a certain self-healing ability of the layer. The value of the friction coefficient calculated from these measurements is about 0.06 at loads up to 100 nN, which is a factor of 2 higher than that observed in water.
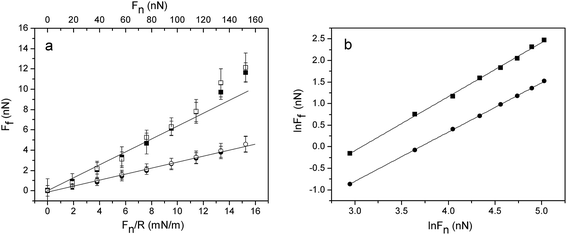 | ||
| Fig. 9 (a) Friction force vs. load for silica surfaces coated with (METAC)m-b-(PEO45MEMA)n after rinsing with water (circles) and with 155 mM NaCl (squares). Filled and unfilled symbols represent data obtained on loading and unloading, respectively. The straight line is a fit to low load data points for illustrating deviations from Amontons' law. (b) The same data plotted on a log–log scale, including the best line fits. | ||
4 Discussion
We devote the discussion section to two aspects that are of importance for the efficiency of a boundary lubricant; (i) the value of the friction coefficient, and (ii) the load bearing capacity. In doing so we compare the performance of the branched brush layer formed by (METAC)m-b-(PEO45MEMA)n with what has been achieved by other polymer layers in aqueous solution.4.1 Friction coefficient
The friction force, Ff, experienced between sliding surfaces is related to the energy dissipated, W, as W = xFf, where x is the sliding distance. Thus, any energy dissipative process that occurs between and within the adsorbed layers during shearing contributes to the measured friction force. In our case there is no adhesion between the (METAC)m-b-(PEO45MEMA)n coated surfaces, and we are thus concerned with load-controlled friction. The friction as a function of load data was analysed using a power law function (see Fig. 9b).| Ff = KFnα | (1) |
It appears that Amontons' rule describes the data well up to a load of 100 nN (corresponding to a pressure of 43 MPa). However, a slightly better fit, lower R2-value, is obtained using a 2-parameter fit (eqn (1)) where the exponent is slightly larger than 1 and the proportionality constant is close to a factor of 2 lower than the friction coefficient deduced from Amontons' rule. We note that it has been suggested that in a single Hertzian asperity contact the friction should scale with the contact area, i.e. with Fn2/3.45 Our data are not consistent with this prediction, but rather suggest that the exponent may be slightly larger than that predicted by Amontons' rule.
One main energy dissipation mechanism between polymer-coated surfaces in the absence of adhesion has been suggested to be due to dragging of polymer chains through the interpenetration zone, i.e. the spatial region where the segment densities emanating from the two opposing layers overlap.46 Thus, if the interpenetration region is small, as can be suggested to be the case when the density of polymer chains is high in a brush layer, one would expect low contribution from this energy dissipative mechanism. The extended bottle-brush structure formed by (METAC)m-b-(PEO45MEMA)n in this investigation is also expected to result in low chain interpenetration due to the branched nature of the brush. This is consistent with the low coefficient of friction of 0.03–0.04 found in this investigation (see Fig. 6 and 9).
The formation and breakage of polymer–surface anchoring points also cause energy dissipation. In this context we note that the magnitude of the friction force between the (METAC)m-b-(PEO45MEMA)n layers is higher in 155 mM NaCl than in pure water (Fig. 9). We suggest that this is a consequence of the decreased electrostatic affinity to the surface, which facilitates shear-induced breakage and reformation of electrostatic segment–surface anchoring points.
When single asperity contacts are considered, Amontons' rule is often slightly modified to take into account deviations observed at low loads, which leads to the relation
| Ff = μFn + C | (2) |
Instead, the deviations from Amontons' rule that we do observe are noticed at high applied loads, as seen in Fig. 9, where the friction force shows a positive deviation from predictions at high loads. We propose that Amontons' rule and the modification of it (eqn (2)) are applicable only as long as the main energy dissipative mechanism is the same. Thus, we suggest that the increase in friction force noted particularly clearly in Fig. 9 at loads above 110 nN is due to the increased importance of a new energy dissipative mechanism. The observation that the up-turn is more significant in 155 mM NaCl than in water suggests that it is due to dragging of polymer chains along the surface, which is facilitated by the weakening of the electrostatic surface affinity at high ionic strength.
The coefficients of friction found for a range of polymer-coated surfaces in water are compared in Table 4. To put these values in perspective, it can be mentioned that the friction coefficient in the synovial joint, a very efficient biotribological system, is in the range 0.001–0.01.3 We note that effective friction coefficients of the order of 0.03, as found in this investigation, are commonly found for surfaces exposing a high density of PEO side chains, whereas significantly higher friction forces are observed when the PEO side chain density is lower (e.g. for PEO45MEMA:METAC-90, see Table 4). Similarly, low friction forces are also observed for layers of the bottle-brush glycoprotein mucin. A common feature of all polymer systems displaying friction forces of 0.04 or below is the presence of a strong steric repulsion that counteracts chain interpenetration. For polymer–surfactant systems, low friction forces have been reported despite the presence of some attractive force contribution.48,49 This has been assigned to water-rich regions within the layers that are sheared without much energy dissipation.49
| System | μ eff | LBC | Comments |
|---|---|---|---|
| (METAC)m-b-(PEO45MEMA)n, silica probe and silica surface | 0.03–0.04 | >50 MPa | This work. Diblock copolymer with one uncharged bottle-brush block and a cationic anchor block without side chains |
| PMMA-b-PSGMA, two mica surfaces11 | 0.0005–0.005 | <1 MPa | Diblock polymer with a hydrophobic block (PMMA) and a polyelectrolytic block (PSGMA). PSGMA tails extend into the aqueous medium to form a polyelectrolyte brush |
| PNIPAAm48-b-PAMPTMA(+)20, silica probe and silica surface48 | 0.5 | — | Diblock copolymer with a cationic anchor block and a poly(N-isopropylacrylamide) block. Low packing density at the surface |
| PS-b-PAA, two mica surfaces50 | 0.3 | 4 MPa | PAA brush built by a diblock copolymer at a controlled surface density by covalent bonds between PS and OH-activated mica surface |
| PEO45MEMA:METAC-10, silica probe and mica surface4 | 0.006 | >30 MPa | PEO45MEMA:METAC-X are random bottle-brush polymers. X represents the percentage of cationic main chain segments, and 100-X corresponds to the percentage of main-chain segments that carry PEO45 side chains |
| PEO45MEMA:METAC-50, silica probe and mica surface4 | 0.03–0.04 | >30 MPa | |
| PEO45MEMA:METAC-90, silica probe and mica surface4 | 0.2 | >30 MPa | |
| AETEC-27-graft-PEO45MEMA, silica probe and silica surface51 | 0.02–0.06 | >35 MPa | Cationic brush-on-brush structure with PEO45 side chains |
| PLL-g-PEO, silica probe and silica surface52 | 0.035 | — | Brush-like copolymer layer built by a cationic main chain having randomly grafted PEO side chains |
| pMPC homopolymer, two mica surfaces12 | 0.0004 | 7.5 MPa | Polyzwitterionic brush |
| Mucin, silica probe and mica surface53 | 0.03 | >40 MPa | Negatively charged bottle-brush glycoprotein |
| Chitosan, silica probe and mica surface53 | 0.13 | >35 MPa | Cationic linear polysaccharide |
| Mucin–chitosan composite layer, silica probe and mica surface53 | 0.4 | >20 MPa | |
| Freely adsorbed and cross-linked chitosan layers, two mica surfaces54 | 0.07 | <1 MPa | Frictional forces increase by cross-linking the chitosan layer |
| 0.5 (cross-linked) | |||
| PLL(20)-g-PEG(2), sodium borosilicate probe and silicon oxide surface55 | 0.20 ± 0.04 | — | Frictional properties of PLL-g-PEG with different PEO chain lengths |
| PLL(20)-g-PEG(5), sodium borosilicate probe and silicon oxide surface55 | 0.199 ± 0.006 | ||
| PLL(20)-g-PEG(10), sodium borosilicate probe and silicon oxide surface55 | 0.162 ± 0.003 |
4.2 Load bearing capacity
Very low friction forces, below the detection limit of the AFM colloidal probe technique, can be obtained provided the surfaces are separated by a water layer that is stabilized by a repulsive surface force, such as a double-layer force48 or a hydration force.56 However, for a double-layer force, the load bearing capacity is limited to low loads, which is one basis for the interest in using polymer layers for achieving low friction in aqueous media.The load bearing capacity of a polymer layer is best defined in terms of pressure, P (rather than load), to allow comparison between different experiments using differently sized surfaces with different Young's moduli. To convert the measured load to a pressure requires the use of a contact mechanics model, and for a small particle in the absence of strong attractive forces the Hertz model is appropriate.57 To this end, the following equations were used to calculate the pressure:
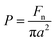 | (3) |
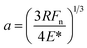 | (4) |
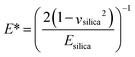 | (5) |
In this study, the maximum load, Fn, was 150 nN, corresponding to a maximum pressure of about 50 MPa. This pressure is 2.5 times higher than has been measured in healthy joints (20 MPa).59 Clearly, the layers formed by (METAC)m-b-(PEO45MEMA)n have a high load bearing capacity. From Table 4, we note that high load bearing capacity has also been achieved for other electrostatically anchored polyelectrolyte layers and for mucin, but for none has the pressure been increased to such high values as in the present investigation. It seems clear that the combination of electrostatic anchoring and a high density of PEO side chains offers both low friction and high load bearing capacity. It should be noted though that the load bearing capacity decreases at high salt concentrations due to weakening of the electrostatic polymer–surface affinity. This important aspect of the lubrication properties of electrostatically anchored surface layers remains to be fully quantified.
5 Conclusions
We have explored the boundary lubrication properties of (METAC)m-b-(PEO45MEMA)n layers adsorbed to silica surfaces. The cationic anchoring block has no PEO side chains and provides a strong attachment to the surface and thereby a high load bearing capacity. In the present study, low friction forces between these layers in pure water were achieved up to a pressure of about 50 MPa, the highest pressure investigated. The low friction force is attributed to strong steric repulsion and low interpenetration between the opposing layers. The friction force increases in high ionic strength solutions, which is suggested to be a consequence of the decreased electrostatic affinity to the surface.The friction force vs. load curves obtained for (METAC)m-b-(PEO45MEMA)n are close to Amontonian, but a small increase above that anticipated from Amontons' rule is observed at high loads, particularly in high ionic strength solutions. This suggests that an additional energy dissipative mechanism becomes important at high loads, and we suggest that this is due to breakage and reformation of electrostatic anchoring points at the surface. Despite this, a low friction force is recovered as the load is decreased again, indicating self-healing properties. This is an advantage of layers formed by a self-assembly process over those formed by chemical grafting where layer disruption is irreversible.
Acknowledgements
XL acknowledges a stipend from the Chinese Scholarship Council (CSC), and PC and MR acknowledge financial support from VR. The project was carried out within the framework of the SSF program “Microstructure, Corrosion and Friction Control”. CV and RM gratefully acknowledge financial support from the Research Council of Lithuania under the project MIP-50/2010. AD enjoys a VINNMER fellowship from VINNOVA.References
- L. Dintenfass, Nature, 1963, 197, 496 CrossRef CAS.
- A. Neville, A. Morina, T. Liskiewicz and Y. Yan, Proc. Inst. Mech. Eng., Part C, 2007, 221, 1223 CrossRef.
- A. Dedinaite, Soft Matter, 2012, 8, 273 RSC.
- T. Pettersson, A. Naderi, R. a. Makuska and P. M. Claesson, Langmuir, 2008, 24, 3336 CrossRef CAS.
- F. Horkay, P. J. Basser, A.-M. Hecht and E. Geissler, J. Chem. Phys., 2008, 128, 135103 CrossRef.
- J. Seror, Y. Merkher, N. Kampf, L. Collinson, A. J. Day, A. Maroudas and J. Klein, Biomacromolecules, 2011, 12, 3432 CrossRef CAS.
- S. Sivan, A. Schroeder, G. Verberne, Y. Merkher, D. Diminsky, A. Priev, A. Maroudas, G. Halperin, D. Nitzan, I. Etsion and Y. Barenholz, Langmuir, 2010, 26, 1107 CrossRef CAS.
- U. Raviv, S. Giasson, N. Kampf, J.-F. Gohy, R. Jerome and J. Klein, Nature, 2003, 425, 163 CrossRef CAS.
- J. F. Joanny, Langmuir, 1992, 8, 989 CrossRef CAS.
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25, 677 CrossRef CAS.
- U. Raviv, S. Giasson, N. Kampf, J.-F. Gohy, R. Jérôme and J. Klein, Langmuir, 2008, 24, 8678 CrossRef CAS.
- M. Chen, W. H. Briscoe, S. P. Armes and J. Klein, Science, 2009, 323, 1698 CrossRef CAS.
- A. Naderi, J. Iruthayaraj, A. Vareikis, R. Makuška and P. M. Claesson, Langmuir, 2007, 23, 12222 CrossRef CAS.
- S. Kinugasa, H. Nakahara, N. Fudagawa and Y. Koga, Macromolecules, 1994, 27, 6889 CrossRef CAS.
- K. Devanand and J. C. Selser, Macromolecules, 1991, 24, 5943 CrossRef CAS.
- E. Mubarekyan and M. M. Santore, Macromolecules, 2001, 34, 7504 CrossRef CAS.
- C. Flood, T. Cosgrove, I. Howell and P. Revell, Langmuir, 2006, 22, 6923 CrossRef CAS.
- Z. Fu and M. M. Santore, Macromolecules, 1998, 31, 7014 CrossRef CAS.
- M. Silvander and M. Silvander, in Lipid and Polymer–Lipid Systems, Springer Berlin, Heidelberg, 2002, vol. 120, p. 35 Search PubMed.
- M. Müller, S. Lee, H. A. Spikes and N. D. Spencer, Tribol. Lett., 2003, 15, 395 CrossRef.
- G. Olanya, E. Thormann, I. Varga, R. Makuška and P. M. Claesson, J. Colloid Interface Sci., 2010, 349, 265 CrossRef CAS.
- S. Pasche, S. M. De Paul, J. Vörös, N. D. Spencer and M. Textor, Langmuir, 2003, 19, 9216 CrossRef CAS.
- Y. Zhou, B. Liedberg, N. Gorochovceva, R. Makuska, A. Dedinaite and P. M. Claesson, J. Colloid Interface Sci., 2007, 305, 62 CrossRef CAS.
- T. Drobek and N. D. Spencer, Langmuir, 2008, 24, 1484 CrossRef CAS.
- P. Linse and P. M. Claesson, Macromolecules, 2009, 42, 6310 CrossRef CAS.
- P. Linse and P. M. Claesson, Macromolecules, 2010, 43, 2076 CrossRef CAS.
- X. Liu, A. Dedinaite, M. Rutland, E. Thormann, C. Visnevskij, R. Makuska and P. M. Claesson, Langmuir, 2012, 28, 15537 CrossRef CAS.
- F. Rico, C. Su and S. Scheuring, Nano Lett., 2011, 11, 3983 CrossRef CAS.
- K. Sweers, K. van der Werf, M. Bennink and V. Subramaniam, Nanoscale Res. Lett., 2011, 6, 270 CrossRef.
- M. Sababi, J. Kettle, H. Rautkoski, P. M. Claesson and E. Thormann, ACS Appl. Mater. Interfaces, 2012, 4, 5534 CAS.
- C. P. Green, H. Lioe, J. P. Cleveland, R. Proksch, P. Mulvaney and J. E. Sader, Rev. Sci. Instrum., 2004, 75, 1988 CrossRef CAS.
- J. E. Sader, J. W. M. Chon and P. Mulvaney, Rev. Sci. Instrum., 1999, 70, 3967 CrossRef CAS.
- T. Pettersson, N. Nordgren, M. W. Rutland and A. Feiler, Rev. Sci. Instrum., 2007, 78, 093702 CrossRef.
- P. Attard, D. J. Mitchell and B. W. Ninham, J. Chem. Phys., 1988, 88, 4987 CrossRef CAS.
- P. Attard, D. J. Mitchell and B. W. Ninham, J. Chem. Phys., 1988, 89, 4358 CrossRef CAS.
- B. Lennart, Adv. Colloid Interface Sci., 1997, 70, 125 CrossRef.
- S. J. Miklavic and S. Marcelja, J. Phys. Chem., 1988, 92, 6718 CrossRef CAS.
- G. E. Fantner, E. Oroudjev, G. Schitter, L. S. Golde, P. Thurner, M. M. Finch, P. Turner, T. Gutsmann, D. E. Morse, H. Hansma and P. K. Hansma, Biophys. J., 2006, 90, 1411 CrossRef CAS.
- T. Hugel and M. Seitz, Macromol. Rapid Commun., 2001, 22, 989 CrossRef CAS.
- C. Ortiz and G. Hadziioannou, Macromolecules, 1999, 32, 780 CrossRef CAS.
- E. Thormann, D. R. Evans and V. S. J. Craig, Macromolecules, 2006, 39, 6180 CrossRef CAS.
- L. M. Grant and F. Tiberg, Biophys. J., 2002, 82, 1373 CrossRef CAS.
- C. E. McNamee, S. Yamamoto, M. Kappl, H.-J. Butt, K. Higashitani, A. Dėdinaitė and P. M. Claesson, J. Colloid Interface Sci., 2011, 364, 351 CrossRef CAS.
- G. Amontons, Mem. Acad. R. A, 1699, 275 Search PubMed.
- Y. Mo, K. T. Turner and I. Szlufarska, Nature, 2009, 457, 1116 CrossRef CAS.
- J. Klein, Annu. Rev. Mater. Sci., 1996, 26, 581 CrossRef CAS.
- B. Bharat, Springer Handbook of Nanotechnology, Springer-Verlag Berlin Heidelberg, Würzburg, 2010 Search PubMed.
- A. Dedinaite, E. Thormann, G. Olanya, P. M. Claesson, B. Nystrom, A.-L. Kjoniksen and K. Zhu, Soft Matter, 2010, 6, 2489 RSC.
- A. Dedinaite, T. Pettersson, B. Mohanty and P. M. Claesson, Soft Matter, 2010, 6, 1520 RSC.
- B. Liberelle and S. Giasson, Langmuir, 2008, 24, 1550 CrossRef CAS.
- T. Krivorotova, R. Makuska, A. Naderi, P. M. Claesson and A. Dedinaite, Eur. Polym. J., 2010, 46, 171 CrossRef CAS.
- M. T. Müller, X. Yan, S. Lee, S. S. Perry and N. D. Spencer, Macromolecules, 2005, 38, 5706 CrossRef.
- T. Pettersson and A. Dėdinaitė, J. Colloid Interface Sci., 2008, 324, 246 CrossRef CAS.
- N. Kampf, U. Raviv and J. Klein, Macromolecules, 2004, 37, 1134 CrossRef CAS.
- S. S. Perry, X. Yan, F. T. Limpoco, S. Lee, M. Müller and N. D. Spencer, ACS Appl. Mater. Interfaces, 2009, 1, 1224 CAS.
- J. Israelachvili, Intermolecular and Surface Forces, Elsevier Inc., London, 2nd edn, 2007 Search PubMed.
- E. Meyer, R. M. Overney, K. Dransfeld and T. Gyalog, Nanoscience: Friction and Rheology on the Nanometer Scale, World Scientific Pub. Co. Inc., Singapore, 2002 Search PubMed.
- M. J. Bamber, K. E. Cooke, A. B. Mann and B. Derby, Thin Solid Films, 2001, 398–399, 299 CrossRef CAS.
- M. Benz, N. Chen, G. Jay and J. Israelachvili, Ann. Biomed. Eng., 2005, 33, 39 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sm27862j |
| This journal is © The Royal Society of Chemistry 2013 |