 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-specific interactions between soluble proteins and lipids induce irreversible changes in the properties of lipid bilayers†
Francesca
Ruggeri‡
a,
Fan
Zhang‡
b,
Tania
Lind
a,
Erica D.
Bruce
b,
Boris L. T.
Lau
b and
Marité
Cárdenas
*a
aInstitute of Chemistry and Nano-Science Center, University of Copenhagen, Universitetparken 5, DK 2100, Copenhagen, Denmark. E-mail: cardenas@nano.ku.dk
bThe Institute of Ecological, Earth, and Environmental Sciences, Baylor University, One Bear Place #97205, Waco, Texas 76798, USA
First published on 12th March 2013
Abstract
Soluble proteins in the extracellular matrix experience a crowded environment. However, most of the biophysical studies performed to date have focused on protein concentrations within the dilute regime (well below the mM range). Here, we systematically studied the interaction of model cell membrane systems (giant unilamellar vesicles and supported lipid bilayers) with soluble globular proteins, bovine serum albumin, hemoglobin and lysozyme at physiologically relevant concentrations. To mimic the extracellular environment more closely, we also used fetal bovine serum as a good representative of a biomimetic protein mixture. We found that regardless of the protein used (and thus of their biological function), the interactions between a model cell membrane and these proteins are determined by their physico-chemical characteristics, mainly their dipolar character (or charged patches). In this paper we discuss the specificity and reversibility of these interactions and their potential implications on the living cells. In particular, we report initial evidence for an additional role of glycolipids in cell membranes: that of reducing the effects of non-specific adsorption of soluble proteins on the cell membrane.
Introduction
The extracellular matrix is a crowded environment where a wide variety of biomacromolecules coexist, including collagen, fibronectin and proteoglycans.1 Earlier it was shown that increasing the protein concentration by using crowding agents can dramatically affect the mobility2–4 and folding5 of proteins. Despite this, dilute conditions are often used in biophysical and biochemical studies precisely with the aim of avoiding intermolecular interactions for simpler data interpretation. For instance, monolayer saturation of globular proteins (e.g., Bovine Serum Albumin, BSA) on solid surfaces typically occurs at ∼50 nM (ref. 6 and 7) and thus little attention has been paid to interfacial processes occurring at higher protein concentrations (>mM) where non-specific8protein–protein and protein–lipid interactions may take place. This is quite surprising given that both Fetal Bovine Serum (FBS) and BSA are commonly used in cell culture media or as blocking agents for molecular diagnostics9 at a concentration of 1–10 volume%.10 Therefore, there is a need for deeper understanding of the protein–lipid behavior in experimental systems that mimic physiological conditions more closely.11 The mM concentration regime for soluble proteins is of high biological relevance given that in blood plasma, a good example of a biological medium, the concentrations of albumin and hemoglobin are ∼0.75 mM (ref. 12) and 0.15 (ref. 13)–2.0 mM (ref. 14) respectively.In this work, we focused on determining the effect of soluble proteins at concentrations closer to their physiological levels on the properties of model cell membranes. These chosen protein concentrations were close to the dilute to semi-dilute regime limit for small globular proteins in solution, which occurs at ∼19 mM to 81 mM (3.4–5.5 nm in diameter), and thus we expect stronger effects due to inter-protein interactions. The semi-dilute regime for a polymer starts at a critical concentration defined as the monomer concentration that is smaller than the solvent concentration but higher than the overlapping concentration.15 As model proteins, we used the most abundant protein in plasma (BSA) and two other globular proteins16 that either carry a similar net charge (hemoglobin)17 or an opposite charge (lysozyme)18 to BSA (for the physico-chemical properties of the proteins used see Table 1). We also used FBS since it represents a good model of a native extracellular protein mixture.
| Proteins used in this work | Molecular weight [kDa] | Zeta-potential [mV] | Hydrodynamic diameter [nm] |
|---|---|---|---|
| a BSA, hemoglobin and lysozyme were measured at 1 mM, and FBS 1% in PBSsolvent. Zeta potential data are reported as average ± standard deviation of six replicates. | |||
| BSA | 66.5 | −11.37 ± 0.96 | 6.8 (ref. 20) |
| Hemoglobin | 64.5 | −2.14 ± 0.90 | 5.5 (ref. 21) |
| Lysozyme | 14.3 | 2.59 ± 0.30 | 4.2 (ref. 20) |
| FBS | NA | −9.05 ± 0.99 | NA |
As model cell membrane systems, we used either giant unilamellar vesicles (GUVs) or supported lipid bilayers (SLBs). Using fluorescence microscopy and GUVs, we assessed major changes in the vesicle structure and/or permeability against a soluble dye19 while quartz crystal microbalance with dissipation (QCM-D) monitoring and SLB allowed us to follow proteinadsorption and the degree of binding reversibility. We used two different types of lipid membranes that were either negatively charged (containing both 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, POPC, and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol, POPG) or neutral (containing POPC only) under pseudo-physiological conditions (pH and salt concentration). Our results indicate that soluble proteins can induce major rearrangements on the model cell membrane at relatively high protein concentrations mainly due to partially reversible binding of proteins to the lipid bilayers at high bulk protein concentrations. In this paper we discuss the specificity and reversibility of these interactions and their potential implications on the living cells. We show initial data suggesting that glycosylation in lipids has the additional role of restricting unspecific protein binding or at least limiting the protein non-specific binding effect on the permeability of lipid bilayers.
Results
Effect of protein co-addition on the permeability of giant unilamellar vesicles
Wide-field fluorescence microscopy experiments were performed to characterize the effects of various proteins on the permeability of lipid membranes against eventual leakage of the soluble dye (Alexa 488) encapsulated in the GUV's lumen. The vesicles carried either a neutral (POPC only) or a net negative charge (POPC/POPG). Furthermore, any major change in the conformation of the lipid bilayers was visualized using DiIC18(5) as a lipid dye. Table 2 presents the minimum protein concentrations required to induce the contained dye leakage in at least 5% of the vesicles within one hour upon exposure to the proteins. Fig. 1A exemplifies the leakage events that occurred for negatively charged vesicles in the presence of hemoglobin. It is clear that all the proteins were capable of inducing leakage from vesicles regardless of their net charge, except for BSA in neutrally charged vesicles. However, the minimum concentration required for dye leakage in 5% of the vesicles significantly differed among both the proteins studied and the net charge of vesicles.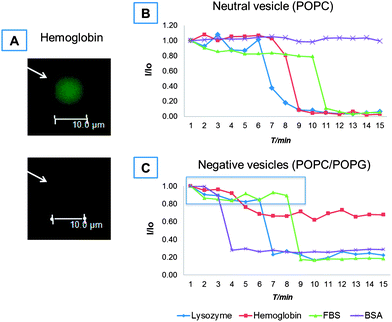 | ||
| Fig. 1 Fluorescence microscopy results for the effect of soluble proteins on GUV permeability against a soluble dye. A representative image of a single vesicle leakage event is given in (A). Representative leakage rate of an individual vesicle expressed as the ratio of real time intensity and initial intensity, I/I0, versus elapsed time on a GUV carrying either a neutral charge (B) or a net negative charge (C) upon exposure to lysozyme, hemoglobin, FBS or BSA. The protein concentrations used in (B) and (C) are those reported in Table 2. | ||
All proteins showed higher affinities against negatively charged than neutral GUVs. For BSA, leakage events were observed for negatively charged vesicles starting from 10 μM, while no leakage events were recorded for neutral vesicles up to 100 μM. Similarly, hemoglobin was more efficient in affecting the permeability of lipid bilayers than BSA regardless of the vesicle charge: a one-fold higher concentration was needed to observe dye leakage for 5% of the charged vesicles while dye leakage occurred at 10 μM for the neutral ones. These results are in agreement with previous studies for hemoglobin,22 where small unilamellar vesicles aggregated and presented peroxidative decomposition in the presence of hemoglobin.
As expected from its overall positive charge,18 lysozyme was more efficient in increasing the permeability against the soluble Alexa dye for the negatively charged vesicles than their neutrally charged counterparts (a one-fold increase in concentration was necessary to affect the permeability of the neutral bilayers). However, the net lysozyme concentration required to induce leakage was one order of magnitude higher than for hemoglobin, even though the latter carried a net negative charge (Table 1). This suggests that there are other mechanisms besides the electrostatic interaction arising from the net charge of proteins and lipids that contributed to the enhanced capability of hemoglobin to change the permeability of lipid bilayers.
FBS was very reactive against GUVs: leakage events were already observable from 0.1% dilution for negatively charged vesicles and from 1% dilution for neutral ones. Finally, at the protein concentrations used, no further modification of the vesicle structure (in terms of bridging, fusion, or vesicle collapse) was observed besides the simple content leakage of Alexa dye except for the case of lysozyme in which vesicle bridging and fusion occurred (ESI, Fig. SI1†), in accordance with the recent findings by Al Kayal et al.23
The kinetics of leakage events were recorded for single vesicles and representative data are shown in Fig. 1B. When neutral vesicles were exposed to hemoglobin and lysozyme, the fluorescence intensity of Alexa dye was fairly stable for ∼6 and ∼8 min respectively until it dropped sharply (1–2 min) to gradually level out at minimal levels. Thus, there seems to be a time threshold for significant dye leakage to occur in the presence of these proteins. However, dye leakage from vesicles occurred immediately after the injection of FBS – although the overall process occurred rather slowly – until reaching 80% of the initial dye intensity (after ∼10 min of mixing). At this point, an abrupt decrease in intensity occurred and, within 1 min, the signal reached minimal levels where it remained stable. In contrast, the dye intensity of vesicles incubated with BSA (at least up to 1 mM) remained constant throughout the experiment.
Interestingly, the negatively charged vesicles were more susceptible to disruption by these proteins despite the electrostatic repulsion expected for negatively charged BSA and hemoglobin (Table 1): not only were the minimum protein (BSA, lysozyme, hemoglobin and FBS) concentrations required for leakage in general one order of magnitude lower (Table 2) but also the intensity drop due to leakage at the individual vesicle level occurred sooner (Fig. 1B and C) in this case. Moreover, all proteins presented two kinetic regimes (slow and fast) during dye leakage. In the slow regime, the initial dye intensity dropped by 20% (expanding over 3–8 min depending on the type of protein). In the fast regime, an additional 60% intensity drop occurred within 1 min for all proteins used except hemoglobin for which only a total of 40% of intensity drop occurred.
Adsorption of proteins to lipid bilayers
We used QCM-D to measure the interfacial properties of proteins on negatively and neutrally charged SLBs. For comparative studies, proteinadsorption on a bare silica substrate was also performed. Silica carried a negative charge under the ionic conditions used (∼0.25 e− nm−2 for SiO2 (ref. 24) as compared to ∼0.54 e− nm−2 for the negatively charged GUVs as estimated from the nominal composition and average mean molecular area for a lipid on a SLB25,26). Fig. 2 summarizes the measured change in frequency (ΔF) and dissipation (ΔD) for neutral and negatively charged SLBs exposed to various protein concentrations. We started by exposing the SLB to the concentrations reported in Table 2 for which the dye leakage reported was observed by fluorescence microscopy. For negatively charged bilayers, no adsorption was observed for lysozyme and hemoglobin, while only a small ΔF (−5 Hz) and ΔD (+1.25 × 10−6) were observed for BSA and FBS. These ΔF and ΔD values correspond to ∼50 weight% of a full protein monolayer (of the size of BSA27) on top of the SLB or ∼77 ng cm−2 based on the Sauerbrey equation (Δm = −CΔfn/n).7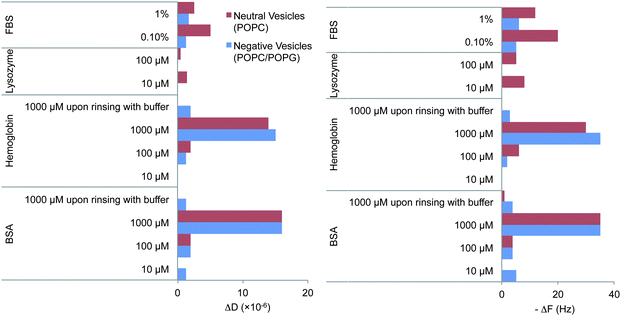 | ||
| Fig. 2 Net absolute changes in frequency (ΔF) and dissipation (ΔD) upon addition of proteins to POPC (neutrally charged) or POPC/POPG (negatively charged) SLB. The figure includes the steady state values upon dilution or extensive rinsing with buffer after exposure to 1 mM BSA and hemoglobin. Red and blue correspond to neutral or negatively charged SLBs respectively. The QCM-D ΔF and ΔD signals typically varied by 2 Hz or 2 × 10−6 dissipation units respectively upon replicating the experiments. Examples of the individual experiments are given in ESI, Fig. SI3–16.† | ||
For neutral membranes, on the other hand, a more pronounced effect was observed for 1% FBS where a steep increase in ΔF (−11 to −14 Hz) was observed. Exposure to 100 μM lysozyme induced a more complex behavior (see Fig. 3) than just simple adsorption (decrease in ΔF and increase in ΔD), where substantial adsorption (ΔF decrease and ΔD increase) was followed by mass removal (ΔF increase and ΔD decrease). Upon reaching steady state conditions, the net ΔF and ΔD values were larger than those for the lipid bilayers prior to lysozyme addition thus indicating that more wet mass than a full lipid bilayer remained bound to the surface. Recently, similar QCM-D responses were obtained for the unfolded equine lysozyme (>3 μM) on negatively charged fluid SLBs (80% PC, 20% PG) while no such effect was observed for native equine lysozyme up to 10 μM,28 in agreement with our results. Indeed at protein concentrations <3 μM, titration of unfolded equine lysozyme leads only to adsorption and no film rearrangement. Thus, here there is also evidence for a threshold surface protein concentration required for major rearrangements of the lipid bilayer.
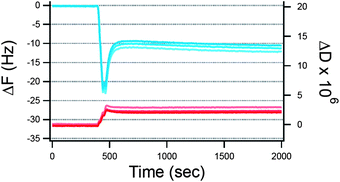 | ||
| Fig. 3 QCM-D signals (ΔF and ΔD in blue and red respectively) for the adsorption of 100 μM lysozyme against time (s) on neutral SLBs. Upon adsorption, a sudden change in the direction of frequency shift indicates the onset of mass removal at t = 500 s after which steady state conditions were reached under continuous flow at 0.1 ml min−1. Overtones shown: 5–7–11. The small overtone spreading indicates the formation of relatively compact and homogeneous layers. | ||
Interestingly, upon increasing the bulk protein concentration a certain critical point is reached where proteins adsorbed on the SLB regardless of the membrane composition or the type of protein as typically expected for non-specific protein binding. The higher the protein concentration, the larger the ΔF and ΔD measured (Fig. 2). Indeed, upon reaching the mM regime (and at comparable concentrations to blood plasma) the ΔD was quite dramatic and there was a considerable spreading of the overtones (ESI, Fig. SI4†). The observed ΔD and ΔF are not related to changes in viscosity of the bulk solution since the exposure of the same solutions to a bare-SiO2 interface gave considerably different results (see Fig. 4) with hemoglobin having a larger capability to self-associate and form multilayers at the SiO2 surface since almost a two times larger change in frequency was observed in this case.
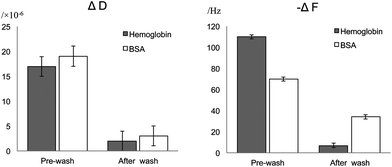 | ||
| Fig. 4 Net changes in frequency (ΔF) and dissipation (ΔD) upon addition of 1 mM BSA or hemoglobin to a bare-SiO2 surface. The figure includes the steady state values upon dilution with excess buffer. | ||
Extensive rinsing with buffer after exposing the lipid bilayers to 1 mM BSA (Fig. SI4 and 7†) or hemoglobin (Fig. SI11 and 12†) was performed to assess the strength of such non-specific protein–lipid binding, see Fig. 2. There was complete protein removal from the neutral SLB since both ΔF and ΔD signals returned to their values prior to protein addition, while only partial removal was achieved for the negatively charged bilayers (the net frequency change after protein removal was −3 Hz) and SiO2 (Fig. 4). The remaining ΔF (−34 and −7 Hz for BSA and hemoglobin respectively on SiO2) and the relatively low D values corresponded well to the reported values for monolayer formation on SiO2, at least for BSA (typically for a small globular protein such as BSA a full monolayer is 160 ng cm−2 or a F change of −45 Hz).27 However, less than a full monolayer remained on the negatively charged bilayers (Fig. 2). Thus, even though a relatively similar extent of proteinadsorption seems to occur upon reaching the mM range on lipid bilayers, the reversibility of the adsorption is hampered on the negatively charged bilayers and corresponded to lower values than on a full monolayer (assuming that lipids are not removed during the washing step).
Since only a small ΔF and ΔD were found for BSA on the SLB under the conditions for which dye leakage occurs from the vesicles (compare Table 2 and Fig. 1), we used commercially available fluorescently labeled (Alexa Fluor® 647) BSA to corroborate the occurrence of BSA adsorption on negatively charged vesicles (Fig. 5). One hour after GUVs exposure to 1 μM labeled-BSA, it was possible to observe an Alexa 647 fluorescent signal around the vesicle membrane, thus suggesting that the albumin indeed adsorbed around the lipid membrane. Note that this very same BSA concentration was below the threshold to induce leakage and thus non-specific proteinadsorption to lipid bilayers occurs readily before any lipid bilayer restructuring occurs. This seems to be in agreement with the two-step mechanism observed for leakage events.
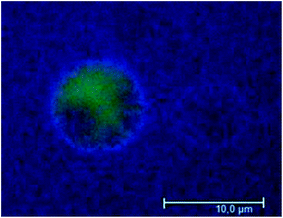 | ||
| Fig. 5 Determination of the effect of labeled BSA on negatively charged GUVs by wide field fluorescence microscopy, one hour after protein addition. Alexa 488 (green) was used to visualize the membrane vesicle's aqueous lumen. Vesicles were exposed to 1 μM Alexa 647-labeled BSA. These results are representative of three different replicates. | ||
Discussion
All proteins studied affected the permeability of giant unilamellar vesicles regardless of the charge of the lipid membrane, except for BSA in neutral vesicles (Fig. 1 and Table 2). The change in permeability is related to proteinadsorption onto lipid membranes, as measured by QCM-D (Fig. 2–4) and fluorescence microscopy (Fig. 5 for BSA only). However, the concentration required for the onset of adsorption (Fig. 1 and Table 2) as well as the extent of adsorption (Fig. 2) was not only dependent on the type of protein but also on the vesicle charge. The relative affinities in terms of the extent of adsorption (net ΔF, Fig. 2) and concentration required to induce content leakage from 5% of the total vesicles imaged (Fig. 1 and Table 2) were not correlated with the zeta potential or apparent surface charge densities of the proteins under the conditions used (Table 1). More intriguingly, a higher affinity in terms of the concentration needed to induce vesicle content leakage was found for similarly charged proteins (hemoglobin) and vesicles (POPG-containing) than for the oppositely charged lysozyme (Table 1) and vesicles (POPG-containing). This seems counterintuitive and surprising due to the overall negative charge of hemoglobin. However, both BSA and hemoglobin possess a highly heterogeneous surface charge distribution,16,17 that gives them a dipolar character with positive patches across the domains (for an extensive recent review on the effects of charge patches on protein interactions see ref. 29). These positive patches are likely to be attracted by the negatively charged head group of the lipids. Indeed, BSA was recently shown to possess affinity towards negatively charged objects30 and to form a full monolayer on SiO2 at around 1 μM.27 Finally, BSA forms a full monolayer at the air–water interface at higher bulk concentrations (1–0 μM).31 Thus, although hydrophobic interactions also contribute to protein–surface interactions, electrostatics between the charged patches of a protein and a charged surface are of longer range. Moreover, these longer range forces are known to dominate the rate of association while short range interactions dominate the dissociation step.32 Indeed, BSA presented similar affinities to lysozyme regardless of having a more negative zeta potential than lysozyme (Table 1). The electrostatic interactions from the net charges of the proteins must anyhow play a role since BSA presented a lower affinity than hemoglobin in accordance with the zeta potential differences between these two proteins (Table 1), which are related to the difference in the protein isoelectric point, 6.8 and 4.7 for hemoglobin and BSA respectively.33,34A closer look at the kinetics of dye leakage indicates that leakage occurs in a two-step mechanism: (1) slow kinetics where no or small change in permeability occurs and (2) fast kinetics where most of the dye leaks within one or two minutes (Fig. 1B and C). This suggests that proteinadsorption occurs readily upon contact with the vesicles leading to some degree of lipid rearrangement, but a critical surface density threshold for the proteins is needed prior to the occurrence of a major leakage. Indeed, the use of labelled BSA corroborated that proteinadsorption occurs prior to any onset of vesicle leakage (Fig. 5). Thus leakage may be related to pore formation viaprotein penetration into the lipid bilayers35,36 or local changes in membrane curvature due to proteinadsorption as suggested earlier for nanoparticle–lipid interactions.37 For instance, initial proteinadsorption occurs without any observable leakage (Fig. 5). For charged membranes, where a slow kinetics leakage step is observed prior to the fast leakage step (Fig. 1) and partially reversible proteinadsorption occurs (Fig. 2), protein penetration into the lipid bilayers in addition to changes in membrane proteins is a more likely scenario. For neutral membranes, on the other hand, leakage occurs only in a fast step and reversible proteinadsorption occurs thus suggesting changes in membrane curvature due to proteinadsorption as a more likely cause for leakage with no penetration into the lipid bilayer occurring in this case.
We measured vast changes in ΔD and ΔF values upon protein exposure to the lipid bilayers that could be related to the formation of soft protein multilayers due to non-specific protein–protein and protein–lipid forces. Interestingly, the lipid membranes were less prone to proteinadsorption than the pristine SiO2 surface since the net ΔF and ΔD were larger in the latter case. In this respect, non-specific inter-protein interactions were detected for a transferrin coated nanoparticle incubated in blood plasma, for which a soft corona of proteins was identified by fluorescent correlation spectroscopy.38 Within this soft corona protein exchange occurred in the timescale of two minutes and faster, showing the highly dynamic nature of such inter-protein non-specific interactions.38 Indeed, the existence of such a soft protein corona arises from typical non-specific protein–protein interactions in the crowded protein environment induced by the higher concentration of proteins at the interface between the solution and any surface. Our results suggest that such reversible, dynamic and “soft” protein–protein interactions also occur in the close vicinity of lipid bilayers and together with non-specific protein–lipid interactions may indeed have an irreversible effect on the lipid bilayer structure, in terms of their permeability against soluble dyes and the colloidal stability of the vesicles.
Lysozyme presents quite a distinct behavior: not only does it induce dye leakage from the vesicles' lumen (Fig. 1), but also induces vesicle aggregation (Fig. SI1†). Moreover, initial lysozyme adsorption on the SLB was followed by a regime of desorption (Fig. 3) and thus a more complex phenomenon than just proteinadsorption occurs in this case. Previously, lysozyme was thought to unfold in contact with lipid membranes upon induction of vesicle fusion23 while it has also been reported that lipid bilayers could induce refolding of denatured lysozyme.28 The QCM-D responses presented in Fig. 3 are indeed typical responses for lipid bilayer restructuring for instance due to molecular insertion and mixed micelle formation.39 Interestingly, unfolded lysozyme due to complexation with oleic acid was found to induce a similar restructuring effect on the negatively charged SLB but such an effect was not observed for native lysozyme below 10 μM.28 Thus, it is clear that the protein concentration is critical since the same mechanism for unfolded lysozyme is reported here as for native lysozyme at 100 μM.
Even though all three proteins tested have very different biological functions, they all presented similar behavior around lipid bilayers in terms of dye leakage and extent of adsorption once a certain protein concentration was reached which was close to the mM range or the semi-dilute regime for small globular proteins. In particular, BSA binds to lysolipids and diacyglycerol,40,41hemoglobin can induce peroxidation of unsaturated lipids under physiological conditions42,43 and lysozyme is a proteolytic enzyme. The latter was also shown recently to induce fusion in negatively charged liposomes under pseudo-physiological conditions.23 In particular, lysozyme is aggressive against the cell membrane of Gram-positive bacteria and this is mainly associated with its anti-bacterial function.44,45 The antibacterial function of lysozyme could, in the light of present results, be indeed related to the actual destabilization of the lipid membrane. Overall, the effect of hemoglobin, BSA and lysozyme on the structure of the lipid bilayers does not seem to be related to their structure but rather to their colloidal properties, mainly in terms of their surface charge density distribution. All proteins are charged and carry a permanent dipole, thus explaining their higher affinity for negatively charged lipid membranes than for neutral ones. Although relatively similar extents of adsorption occurred upon reaching the mM concentration range at least for similarly charged membranes (Fig. 2), the reversibility of the non-specific proteinadsorption was hampered on the negatively charged bilayers only, at least for BSA and hemoglobin (Fig. 2 and 4). Thus, the stronger electrostatic interactions between the proteins and the lipids also induced non-reversibility effects. Thus, it is clear that besides favourable electrostatic interactions between the charged patches of the proteins and the lipid bilayer that drive the protein–lipid binding there are other short range interactions (van der Waals, hydrogen bonds, hydrophobic interactions and salt bridges) that decrease the protein dissociation from the lipid bilayers. This is a typical behaviour observed for proteins and polyelectrolytes in general as profoundly revised recently by Kayitmazer et al.29
For FBS, on the other hand, leakage events were observable starting from 0.1% dilution for negatively charged GUVs and from 1% dilution for neutral GUVs (Table 2 and Fig. 1). The concentration commonly used46 in cell culture studies is 10% dilution by volume of this media, and thus FBS addition should induce proteinadsorption on cell membranes given that conditions are kept similar to the ones hereby used (cell versus vesicle densities). However, FBS or BSA addition does not induce cell lysis in cell cultures. Indeed, we used protein concentrations close to or within their physiological values. Thus, none of these proteins should be able to affect the cell membrane structure in vitro or in vivo. Therefore, the cell must possess other mechanisms to protect itself from the action of soluble proteins in the mM concentration regime. The head groups of glycosylated lipids are highly hydrated and are known to actively protect the lipid membrane from desiccation47–49 and peroxidation.50 We hypothesize that glycolipids have yet another biological function51 related to the hindrance of soluble protein binding. This hypothesis is supported by the fact that sugars are used in surface coating for preventing non-specific protein binding.52 Indeed, preliminary experiments show that the use of phosphatidyl inositol (PI) instead of phosphatidylglycerol (PG) at the same molar ratio (and thus the same surface charge density) decreases the effect of protein binding on the permeability of lipid membranes since no leakage event was recorded for titration with up to 50 μM BSA, see Fig. 6. However, further experimentation with various conditions is needed to corroborate this hypothesis as a general effect for other proteins.
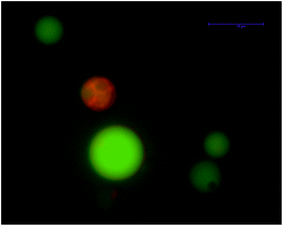 | ||
| Fig. 6 No significant leakage was observed by wide field fluorescence microscopy upon exposing negatively charged GUVs made of phosphatidylinositol (25 mol%) one hour after 50 μM BSA addition. Alexa 488 (green) was used to visualize the membrane vesicle's aqueous lumen. DilC18(5) was used to mark the lipid bilayer (red). These results are representative of three different replicates. | ||
Experimental
Materials
POPG, POPC and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-n-cap biotinyl (DOPE-biotin) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) while 1,2-dipalmitoyl-phosphatidylinositol (DPPI) was purchased from Larodan Fine Chemicals AB. All lipids were used without further purification. The dyes DiIC18(5) (1,1′-dioctadecyl-3,3,3′,3′-tetramethy-lindodicarbocyanineperchlorate) and Alexa Fluor® 488 hydrazide (Alexa) were used as received from Invitrogen (Paisley, UK). Alexa Fluor® 647 conjugate was purchased from Invitrogen (Paisley, UK), dispersed in PBS to a concentration of 100 μM, and stored at −20 °C until use.Preparation and characterization of vesicles
Two types of GUVs were prepared by mixing the two lipids, POPG and POPC, at the molar ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 and 0:1, respectively. Based on the pKa of the head groups at physiological pH, POPC/POPG vesicles carried a negative charge, while POPC carried a net charge of zero.53 Additionally, DPPI was mixed with POPC at a molar ratio of 1:3 to give vesicles with the same charge as those made using POPG but carrying an inositol group instead of glycerol.
:3 and 0:1, respectively. Based on the pKa of the head groups at physiological pH, POPC/POPG vesicles carried a negative charge, while POPC carried a net charge of zero.53 Additionally, DPPI was mixed with POPC at a molar ratio of 1:3 to give vesicles with the same charge as those made using POPG but carrying an inositol group instead of glycerol.
For the leakage studies, both types of vesicles contained 1% molar ratio of the lipiddye DiIC18(5) and 0.5% molar ratio of DOPE-biotin, to tether the vesicles to the surface of the sample chamber. The lipids were dissolved in chloroform at appropriate ratios and the solvent was evaporated under vacuum for at least one hour. The films were then rehydrated with 10 μM Alexa 488 hydrazide in sorbitol solution, followed by overnight water bath incubation at 37 °C. This procedure enabled the vesicles to carry DiIC18(5) in the lipid bilayers while the soluble Alexa 488 was confined to the vesicle lumen. The final vesicle solutions were stored at 4 °C and used within a week.
For QCM-D experiments, small unilamellar vesicles (SUVs) were prepared at the same molar ratios of the two lipids as stated above. However, the vesicles did not contain DiIC18(5) or DOPE-biotin in this case. The POPC/POPG vesicles were rehydrated in 2 mM CaCl2 to facilitate the formation of the supported lipid bilayer by bridging the negatively charged vesicles with the SiO2 surface, while POPC vesicles were simply rehydrated in Milli-Q water. Vesicles were subjected to two to five minutes of sonication until the solution was clear and transparent before use.
Preparation and characterization of proteins
Bovine serum albumin (BSA) with a purity of above 98%, human blood hemoglobin (hemoglobin), fetal bovine serum (FBS) and chicken egg white lysozyme (lysozyme) were purchased from Sigma-Aldrich (Denmark) and used without further purification. The proteins were first dispersed in phosphate buffer saline (pH 7.4, ionic strength 127 mM) and diluted further in the same buffer to the experimental concentrations. In this study, protein concentrations for BSA, hemoglobin and lysozyme ranged from 1 μM to 1 mM, while FBS was studied at 0.1%, 1% and 2% volume ratios. The zeta potential of each type of protein was determined by electrophoretic mobility measurements using a Malvern Zetasizer NS (Worcestershire, U.K.), see Table 1. In this case, the samples were prepared by dispersing the proteins in PBS to a final concentration of 1 mM, FBS was diluted in PBS to 1% by volume; and six measurements were performed on each sample at 25 °C.Description of microscopy experiments
The sample chambers were incubated with 1.0 g l−1 BSA-biotin–BSA solution (1:10, volume ratio), 0.025 g l−1 streptavidin and PBS, respectively, at ambient temperature for 10 minutes, plus rinsing with PBS five times. Fluorescent vesicles were added to the chamber to achieve a final concentration of 0.01 mg ml−1. The vesicles were allowed to settle and stabilize at the bottom of the chamber for 30 minutes before injection of proteins into the solution. After exposure to proteins, the behaviors of GUVs were monitored for up to 60 minutes on a Leica AF6000LX wide-field microscope (Wetzlar, Germany). For each dye leakage experiment, 10 to 15 locations were imaged with approximately 20 vesicles in each location, which made up to 200 to 300 vesicles in every study. The fluorescence of Alexa was excited at λ = 488 nm with an argon laser and emission was captured at λ = 491–563 nm. The lipid dye, DiIC18(5), was excited at λ = 633 nm and recorded at λ = 640–700 nm. A mercury lamp with filter cubes EGF cube 49002 ET and EC5 cube 49006 ET (Chroma Technology Corp, Bellows Falls, USA) was used to excite Alexa and DiIC18(5) respectively. For each experimental setup, illumination, intensity and exposure time were optimized for signal collection. The integrity of vesicles was checked in a buffer solution during control experiments, without exposure to proteins, to ensure their stability against mechanical stress, and no sign of dye leakage was observed. Each set of experiments was repeated at least three times.
Description of QCM-D experiments
QCM-D (quartz crystal microbalance with dissipation monitoring) is an acoustic technique for measuring (1) the change of wet mass per unit area by sensing ΔF of a quartz crystal resonator and (2) the viscoelastic properties of the adsorbed film by sensing the energy dissipation (D). A decrease in resonance frequency is an indicator of an increase in adsorbed mass on the sensor surface while an increase in dissipation is related to a decrease in rigidity of the adsorbed film. In this study, we used a Q-Sense E4 system (Gothenburg, Sweden). All SiO2 sensors were soaked in 2% Hellmanex for 10 minutes and washed by 15 rinsing cycles of water and 96% ethanol, followed by an UV-ozone cleaning process for another 10 minutes to remove any possible contamination layer on the sensor surface. Each measurement was performed at 25 °C when the frequency changes were within 0.5 Hz. 5% of sodium dodecyl sulfate (SDS) was applied for cleaning, and 96% ethanol was used to remove bubbles in the tubing when necessary. Upon obtaining a stable signal, a vesicle solution (200 μg ml−1) was injected at a rate of 100 μl min−1. After the formation of SLBs, PBS was pumped into the system to wash off excess lipids before the injection of proteins. A series of protein titrations were then performed, starting from 1 μM to 1 mM for all proteins except FBS for which 0.1% to 2% of FBS was used.Conclusions
Non-specific proteinadsorption has now been studied in view of its effect on the structure of lipid bilayers in terms of vesicle colloidal stability and lipid bilayer permeability. We found that all proteins studied (bovine serum albumin, chicken egg lysozyme, bovine hemoglobin and bovine fetal serum) were able to induce leakage from lipid vesicles regardless of their surface charge (negative or neutral) expect for albumin on neutral vesicles at least up to 1 mM. The protein concentrations required for leakage to occur were well above the μM range where strong inter-protein interactions occur. At the same time, this is the concentration regime that is physiologically relevant. Evidently, vast soluble proteinadsorption on the cell membranes does not occur in vivo. We propose that the cell possesses other mechanisms to defend itself from this type of non-specific proteinadsorption among which the glycosylated coatings introduced by lipids and membrane proteins could be a major parameter in play.Acknowledgements
We thank Karen Martinez for providing access to the wide field microscope and Thomas Günter Pomorski for fruitful discussions. Moreover, the authors gratefully acknowledge financial support from “Center for Synthetic Biology” at Copenhagen University funded by the UNIK research initiative of the Danish Ministry of Science, Technology and Innovation, and C. Gus Glasscock, Jr, endowed fund for excellence in environmental sciences in the College of Arts and Sciences by Baylor University, USA.References
- M. Iwata and S. S. Carlson, J. Neurosci., 1993, 13, 195–207 Search PubMed.
- Y. Y. Kuttner, N. Kozer, E. Segal, G. Schreiber and G. Haran, J. Am. Chem. Soc., 2005, 127, 15138–15144 CrossRef CAS.
- D. S. Banks and C. Fradin, Biophys. J., 2005, 89, 2960–2971 CrossRef CAS.
- M. Weiss, M. Elsner, F. Kartberg and T. Nilsson, Biophys. J., 2004, 87, 3518–3524 CrossRef CAS.
- S. Mukherjee, M. M. Waegele, P. Chowdhury, L. Guo and F. Gai, J. Mol. Biol., 2009, 393, 227–236 CrossRef CAS.
- C. Larsson, M. Rodahl and F. Hook, Anal. Chem., 2003, 75, 5080–5087 CrossRef.
- P. M. Wolny, J. P. Spatz and R. P. Richter, Langmuir, 2010, 26, 1029–1034 Search PubMed.
- A. V. Kiselev, Discuss. Faraday Soc., 1965, 205–218 Search PubMed.
- E. Pozio, L. Sofronic-Milosavljevic, M. A. Gomez Morales, P. Boireau and K. Nöckler, Vet. Parasitol., 2002, 108, 163–178 Search PubMed.
- C. Incorporated, in http://www.corning.com/lifesciences.
- A. H. Elcock, Curr. Opin. Struct. Biol., 2010, 20, 196–206 CrossRef CAS.
- R. L. Lundblad, Internet J. Genomics Proteomics, 2005, 1 Search PubMed , ISSN: 1540–2630.
- C. S. Broberg, A. R. Jayaweera, G. P. Diller, S. K. Prasad, S. L. Thein, B. E. Bax, J. Burman and M. A. Gatzoulis, Am. J. Cardiol., 2011, 107, 595–599 Search PubMed.
- L. Detivaud, E. Nemeth, K. Boudjema, B. Turlin, M. B. Troadec, P. Leroyer, M. Ropert, S. Jacquelinet, B. Courselaud, T. Ganz, P. Brissot and O. Loreal, Blood, 2005, 106, 746–748 Search PubMed.
- M. Adam and M. Delsanti, Macromolecules, 1985, 18, 1760–1770 CrossRef CAS.
- Q. Shi, Y. Zhou and Y. Sun, Biotechnol. Prog., 2005, 21, 516–523 Search PubMed.
- S. X. Wang, K. C. Pandey, J. R. Somoza, P. S. Sijwali, T. Kortemme, L. S. Brinen, R. J. Fletterick, P. J. Rosenthal and J. H. McKerrow, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11503–11508 CrossRef CAS.
- D. P. Sun, D. I. Liao and S. J. Remington, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 5361–5365.
- A. Akesson, C. V. Lundgaard, N. Ehrlich, T. G. Pomorski, D. Stamou and M. Cardenas, Soft Matter, 2012, 8, 8972–8980 RSC.
- F. L. González Flecha and V. Levi, Biochem. Mol. Biol. Educ., 2003, 31, 319–322 CrossRef CAS.
- S. Papadopoulos, K. D. Jurgens and G. Gros, Biophys. J., 2000, 79, 2084–2094 CrossRef CAS.
- J. Szebeni, H. Hauser, C. D. Eskelson, R. R. Watson and K. H. Winterhalter, Biochemistry, 1988, 27, 6425–6434 Search PubMed.
- S. N. Tamer Al Kayal, E. Russo, D. Berti, M. Bucciantini, M. Stefani and P. Baglioni, Soft Matter, 2012, 8, 4524–4534 RSC.
- G. H. Bolt, J. Phys. Chem., 1957, 61, 1166–1169 CrossRef CAS.
- A. Akesson, T. Lind, N. Ehrlich, D. Stamou, H. Wacklin and M. Cardenas, Soft Matter, 2012, 8, 5658–5665 RSC.
- A. Akesson, T. K. Lind, R. Barker, A. Hughes and M. Cardenas, Langmuir, 2012, 28, 13025–13033 Search PubMed.
- G. Olanya, E. Thormann, I. Varga, R. Makuska and P. M. Claesson, J. Colloid Interface Sci., 2010, 349, 265–274 CrossRef CAS.
- S. B. Nielsen, K. Wilhelm, B. Vad, J. Schleucher, L. A. Morozova-Roche and D. Otzen, J. Mol. Biol., 2010, 398, 351–361 CrossRef CAS.
- A. B. Kayitmazer, D. Seeman, B. B. Minsky, P. L. Dubin and Y. Xu, Soft Matter, 2013, 2553–2583 Search PubMed.
- B. Jachimska and A. Pajor, Bioelectrochemistry, 2012, 87, 138–146 Search PubMed.
- C. Ybert and J. M. di Meglio, Langmuir, 1998, 14, 471–475 CrossRef CAS.
- T. Selzer, S. Albeck and G. Schreiber, Nat. Struct. Biol., 2000, 7, 537–541 CrossRef CAS.
- T. Shiomi, M. Matsui, F. Mizukami and K. Sakaguchi, Biomaterials, 2005, 26, 5564–5571 CrossRef CAS.
- S. R. Bellara, Z. F. Cui and D. S. Pepper, Biotechnol. Prog., 1997, 13, 869–872 CrossRef CAS.
- G. Fuertes, D. Gimenez, S. Esteban-Martin, A. Garcia-Saez, O. Sanchez and J. Salgado, Adv. Exp. Med. Biol., 2010, 677, 31–55 Search PubMed.
- M. J. Zuckermann and T. Heimburg, Biophys. J., 2001, 81, 2458–2472 CrossRef CAS.
- A. Akesson, T. K. Lind, R. Barker, A. Hughes and M. Cardenas, Langmuir, 2012, 28, 13025–13033 Search PubMed.
- S. Milani, F. B. Bombelli, A. S. Pitek, K. A. Dawson and J. Radler, ACS Nano, 2012, 6, 2532–2541 Search PubMed.
- A. Mechler, S. Praporski, K. Atmuri, M. Boland, F. Separovic and L. L. Martin, Biophys. J., 2007, 93, 3907–3916 CrossRef CAS.
- D. M. Ojcius and J. D. Young, Mol. Immunol., 1990, 27, 257–261 Search PubMed.
- H. Ahyayauch, G. Arana, J. Sot, A. Alonso and F. M. Goni, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 701–707 CrossRef CAS.
- W. S. Pietrzak and I. F. Miller, Biomater., Artif. Cells, Artif. Organs, 1989, 17, 563–581 Search PubMed.
- J. Szebeni, C. C. Winterbourn and R. W. Carrell, Biochem. J., 1984, 220, 685–692 Search PubMed.
- V. J. Iacono, B. J. MacKay, S. DiRienzo and J. J. Pollock, Infect. Immun., 1980, 29, 623–632 Search PubMed.
- R. Cegielska-Radziejewska, G. Lesnierowski and J. Kijowski, Eur. Food Res. Technol., 2009, 228, 841–845 Search PubMed.
- J. Choi, J. H. Chung, G. Y. Kwon, K. W. Kim, S. Kim and H. Chang, Cell Tissue Banking, 2012, 1–10 Search PubMed.
- C. W. Harland, Z. Botyanszki, D. Rabuka, C. R. Bertozzi and R. Parthasarathy, Langmuir, 2009, 25, 5193–5198 CrossRef CAS.
- S. B. Leslie, E. Israeli, B. Lighthart, J. H. Crowe and L. M. Crowe, Appl. Environ. Microbiol., 1995, 61, 3592–3597 CAS.
- S. J. Halperin and K. L. Koster, J. Exp. Bot., 2006, 57, 2303–2311 Search PubMed.
- I. Varani, A. Terzaghi, L. Donati, M. Marazzi, M. Masserini and G. Tettamanti, Arch. Dermatol. Res., 1994, 286, 481–483 Search PubMed.
- A. Varki, Glycobiology, 1993, 3, 97–130 CrossRef CAS.
- G. Jogikalmath, US Pat., US 2008/0213910 application publication, 2008.
- A. Dickey and R. Faller, Biophys. J., 2008, 95, 2636–2646 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: QCM-D graphs with raw data and fluorescence microscopy leakage images. See DOI: 10.1039/c3sm27769k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |