 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of degree of polymerization on intra- and interchain micellization of a tail-type cationic polysoap†
Paul A.
FitzGerald
,
David M.
McDonald
and
Gregory G.
Warr
*
School of Chemistry, F11, The University of Sydney, 2006, Australia. E-mail: gregory.warr@sydney.edu.au; Fax: +612 9351 3329; Tel: +612 9351 2106
First published on 23rd January 2013
Abstract
We have used Reversible Addition Fragmentation chain Transfer (RAFT) to polymerize the T-type surfactant monomer α,ω-methacryloylundecyltrimethylammonium bromide (MUTAB) to various degrees of polymerization, and thereby investigate how its self-assembly is affected. Small-angle neutron scattering (SANS) shows that the interchain aggregation into micelles with an approximately constant number of MUTAB monomer equivalents occurs at low degrees of polymerization, but that micelle elongation occurs when the degree of polymerization exceeds a critical value. In this regime interchain aggregation gives way to intrachain assembly into unimolecular or “unimer” micelles. As with conventional cationic surfactant solutions, addition of salicylate produces long, worm-like micelles containing many amphiphilic polymer chains at all degrees of polymerization. Oscillatory rheology reveals a transition from scission- to reptation-dominated relaxation as increasing polymer chain length also increases the distance between potential scission points. The measured relaxation times lie in the range of hundredths to a few seconds – thus demonstrating the rapidly equilibrating nature of these micellar systems even at the highest degrees of polymerization achieved.
Introduction
Attempts to polymerize assemblies of surfactant monomers (surfmers) are found throughout the literature.1–14 Typically the aim is to kinetically trap some property of the original micelles, such as size1 or morphology.15,16 This is rarely achieved, and most studies note changes due to the polymerization such as micelle growth.15,16 However, it is possible to retain some features of the original micelle such as the cross-sectional radius,12,13 and the polymerization is also often found to impart stability to the polymerized micelles against dilution and changes in temperature.15,16Apart from demonstrated stability to temperature and dilution, there are surprisingly few studies on the stability of polymerized micelles to other classic micelle morphology modifies such as salt or additional surfactant. Most authors shy away from drawing firm conclusions about the thermodynamic nature of the polymerized structures, whether the final product is a polymer particle or an equilibrium polysoap micelle, or whether they contain one or many polysoap chains.15,16 Further, because traditional polymerization methods afford little in the way of controlling polymerization, it has previously not been possible to study the influence of molecular weight on the final structures formed. In this work we use Reversible Addition–Fragmentation chain Transfer (RAFT) to synthesize a series of polysoaps with varying degree of polymerization, and elucidate the nature and relationship of both polymerized micelles and self-assembled polysoaps as a function of molecular weight.
Recently we have shown that micelles of the classic tail-polymerizable surfactant monomer MUTAB (T-type surfmer, Fig. 1) remain in thermodynamic equilibrium throughout the in situ polymerization process.17–19 It forms small spheroidal micelles before polymerization (Nagg = 42) and larger spheroids after polymerization with a four-fold increase in length, but only a minor increase in the cross-sectional radius. At intermediate polymer conversions, rod-like micelles form with lengths of more than 1500 Å, which collapse into polymerized spheroids as the monomer is consumed. These fully polymerized micelles are stable to both dilution and temperature changes, but remain in dynamic equilibrium in that they undergo facile and extensive structural rearrangement in response to the addition of, for example, salt or monomeric surfactant,14,19 or a change in temperature.14
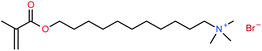 | ||
| Fig. 1 Tail polymerizable surfactant monomer (surfmer) MUTAB. | ||
In this work we compare the structure and properties of in situ polymerized aqueous MUTAB micelles with MUTAB polysoaps prepared ex situ using RAFT to control the degree of polymerization. We also compare the behaviour of these polymer chains in non-self-assembling and self-assembling solvents, drawing a distinction between inter- and intrachain association to form unimer micelles. Finally, we examine long, worm-like micelles made from polymerized MUTAB with added salicylate, and show how polymerization significantly affects solution rheology through micelle dynamics.
Experimental section
MUTAB was synthesized as described previously.20 The micellar polymerization was performed using UV polymerization at 2× CMC (∼1.7 wt% in D2O), under the same conditions as described previously. Conductivity measurements (see ESI, Fig. S1†) confirm that the polymerized MUTAB micelles do not disassemble on dilution to at least 1/20th of the CMC of monomeric MUTAB, and indicates that micelle counterion binding is similar before and after polymerization.A series of polymerized MUTAB samples were synthesised to varying degrees of polymerization, n, using the RAFT controlled polymerization technique.21,22 RAFT polymerizations of MUTAB were conducted on approximately 30 wt% MUTAB samples in methanol by thermal initiation at 70 °C for 16 hours using azoisobutyrylnitrile (AIBN) initiator and cyanopropyldithiobenzene (CPDB)21,22 RAFT control agent in the mole ratio 0.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:n (AIBN:CPDB:MUTAB) where n = 20, 40, 90, 200 and 500 is the desired degree of polymerization. The polymer mixture was precipitated in ether to remove any residual monomer and dried under vacuum before being redissolved in either D2O or deuterated methanol for SANS experiments. The RAFT polymers are capped by the small hydrophobic end groups of cyanopropyl and dithiobenzene, which add a negligible hydrophobic contribution to the polymeric backbone.
:1:n (AIBN:CPDB:MUTAB) where n = 20, 40, 90, 200 and 500 is the desired degree of polymerization. The polymer mixture was precipitated in ether to remove any residual monomer and dried under vacuum before being redissolved in either D2O or deuterated methanol for SANS experiments. The RAFT polymers are capped by the small hydrophobic end groups of cyanopropyl and dithiobenzene, which add a negligible hydrophobic contribution to the polymeric backbone.
The actual degree of polymerization for the larger molecular weights was measured using SANS on 5 mg mL−1 samples in deuterated methanol brine (containing 0.1 M NaCl to screen any electrostatic interactions), and aqueous micellar solutions in D2O under various solution conditions (see Results and discussion).
Small-angle neutron scattering (SANS) was performed on the 18 m SANS instrument at the HANARO reactor at the Korean Atomic Energy Research Institute (KAERO) and the NG3 SANS instrument at the NIST Center for Neutron Research (NCNR) at Gaithersberg MD,23 using 2 mm path length cells with 5.14 Å (HANARO) or 6.0 Å (NCNR) neutrons collected on a 128 × 128 pixel, 640 × 640 mm2 detector at 1.3 m and 9 m sample to detector distances, giving a combined q range of 0.008–0.3 Å−1. Data reduction and analysis was performed in Igor Pro (Wavemetrics Inc., version 6.22A) using the NIST reduction and analysis macros (version 4.0).24
SANS data for molecular weight determination was collected at an optimized concentration (∼5 mg mL−1), which was large enough to give sufficient contrast yet low enough to exclude almost all polymer–polymer interactions. The optimized concentration was determined by collecting SANS data for the micelle polymerized sample at several concentrations between 1.0 and 20 mg mL−1 and then establishing the maximum concentration below which the normalized scattering (i.e. I(q)/c) overlay each other within experimental error.
Small-angle X-ray Scattering (SAXS) was performed on an Anton Paar SAXSess using 1 mm diameter quartz sample capillaries, and a 1 cm collimated line. Scattering was collected on image plates and desmeared using Otto Glatter's GIFT program to obtain the scattering intensity at q = 0 (i.e. I(0)).25
Rheology data was collected at 25 °C on an Anton Paar MCR 302 rheometer using a parallel plate geometry with a 50 mm diameter and 0.5 mm gap. Samples consisted of the polymerized MUTAB in D2O with sodium salicylate (0.82 mol%) to induce micelle elongation and network formation.
Results and discussion
M w and Rg of polyMUTAB in methanol
SANS patterns for the polymerized MUTAB samples in deuterated methanol – with 0.1 M NaCl added to screen any electrostatic interactions – are shown in Fig. 2. For ease of comparison, all intensities are normalized by concentration (w/v) and the backgrounds subtracted. The low q data was fit using the Zimm approximation,26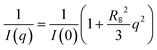 | (1) |
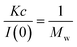 | (2) |
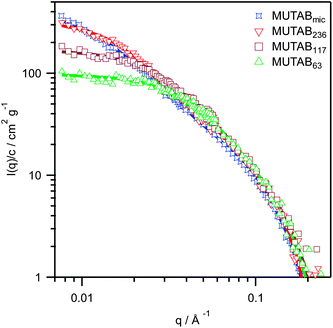 | ||
| Fig. 2 SANS patterns for polymerized MUTAB samples in deuterated methanol + 0.1 M NaCl with degrees of polymerization of 63 (triangles), 117 (squares), 236 (upside down triangles) and the micelle polymerized sample (n = 275, stars). Fits are to the flexible cylinder model.30,31 | ||
The degree of polymerization, n, was calculated by dividing the measured Mw by the monomer molecular weight (MUTA+, 298 g mol−1) and is given in Table 1 with corresponding Rg values from eqn (1) (full fit values given in Table S1, ESI†). The degree of polymerization could not be measured for the shorter polymers (target values of 20 and 40) using SANS and were instead measured with SAXS, using MUTAB63 from SANS as a secondary standard.
| Target n | MUTAB | 20 | 40 | 90 | 200 | 500 | Micellar |
|---|---|---|---|---|---|---|---|
| a Uncertainties are approximately ±5% for lengths (1–10 Å) and ±20% for volume fractions. | |||||||
| In d-methanol | |||||||
| Zimm analysis | |||||||
| Measured n | 1 | 14 | 28 | 63 | 117 | 236 | 275 |
| R g/Å | — | — | — | 40 | 64 | 108 | 132 |
| Flexible rod model | |||||||
| Scale | — | — | — | 0.0012 | 0.0012 | 0.0012 | 0.0009 |
| Contour Length/Å | — | — | — | 212 | 393 | 584 | 811 |
| Kuhn Length/Å | — | — | — | 37 | 39 | 82 | 98 |
| Radius/Å | — | — | — | 14 | 13 | 15 | 15 |
| In D 2 O | |||||||
| Fits from prolate spheroid model | |||||||
| Vol. frac. | 0.011 | 0.012 | 0.017 | 0.019 | 0.02 | 0.019 | 0.017 |
| R a/Å | 26 | 26 | 27 | 36 | 54 | 79 | 101 |
| R b/Å | 14 | 17 | 18 | 19 | 18 | 19 | 19 |
| N agg,mon eq. | 42 | 65 | 69 | 106 | 155 | 228 | 305 |
| N agg | 42 | 4.7 | 2.4 | 1.7 | 1.3 | 1.0 | 1.1 |
The experimental degrees of polymerization for RAFT and the micelle polymerized MUTAB, and their corresponding radii of gyration in methanol brine are listed in Table 1. Degrees of polymerization are lower than the target values due to termination prior to complete conversion, with unreacted monomer removed during the precipitation step. Fig. 3 shows that the radii of gyration exhibit a power law dependence of Rg ∼ n0.81, which is intermediate between the n0.6 dependence expected for long excluded-volume polymers,32 and the n1.0 expected for rigid rods.32
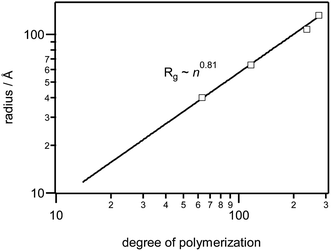 | ||
| Fig. 3 Polymer chain radii of gyration in d-methanol brine. | ||
The SANS patterns in methanol were fit to a flexible cylinder model30,31 as shown in Fig. 2, with the best-fit parameter values given in Table 1. The model provides good fits over the entire q range, and shows that there are only 5 to 10 Kuhn segments, each 40 to 100 Å in length, per polymer chain. These are almost an order of magnitude larger than typical methacrylates, e.g. 7.2 Å for poly(methyl methacrylate) in toluene,33 probably due to electrostatic repulsion between monomers along the polymer chain. Indeed, the 6.2 Å Debye length in a 0.1 M NaCl methanol solution at 25 °C is large enough to screen interactions between polymer chains 100–200 Å apart, but not between adjacent charges along the chain, which are only 3–9 Å apart. The large Kuhn lengths also suggest that the observed Rg ∼ n0.81 scaling behaviour is actually the chains behaving somewhere between rigid rods and an excluded volume polymer.
polyMUTAB micelle morphologies in D2O
SANS patterns for 2 wt% RAFT-polymerized MUTAB solutions in D2O at various degrees of polymerization, n, are shown in Fig. 4, together with both micelle polymerized and monomeric MUTAB at 2xCMC of the monomer (1.7 wt%). The data were best fit as prolate spheroids34 interacting through a screened Coulomb interaction,35,36 with key fit values given in Table 1 (full fit values in Table S2, ESI†).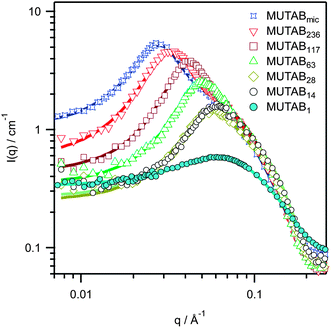 | ||
| Fig. 4 SANS patterns of micellar solutions of 2 wt% MUTABn in D2O. Solid lines show with fits to a prolate spheroid model with screened Coulomb interaction. Monomer, MUTAB1, and micelle polymerized, MUTABmic, at 2 × CMC = 1.72 wt% are also shown.18 | ||
The cross-sectional radius, Rb = 18 ± 1 Å, is practically independent of n, (apart from monomeric MUTAB, which has been shown previously to have a smaller radius due to “hair-pinning” of the tails to allow the polar methacrylate group closer to the micelle surface).17,18 The polymerized radius is consistent with fully extended non-polar tails, which are indifferent to the polymethacrylate backbone and instead dictated by packing constraints. At low n the long axis, Ra, and hence the micelle size, is independent of the degree of polymerization i.e. Ra = 26 ± 1 Å at n = 1, 14 and 28. However, for larger n the micelles elongate uniaxially, rapidly approaching the Ra ∼ n1.0 dependence expected for rigid rods.
The micelle aggregation numbers in monomer equivalents, Nagg,mon eq., obtained by dividing the volume of the spheroidal micelles by the volume of a MUTA+ monomer, (496 Å3), is insensitive to n up to about 28-mers, but thereafter increases markedly. The reason for this is readily seen by considering the number of actual polyMUTAB units in each micelle, Nagg. Both values are listed in Table 1.
At low n, each micelle is formed by the association of several polymer chains (i.e. Nagg > 1), and the micelle aggregation number is determined by packing constraints. However, once the degree of polymerization of an individual polyMUTAB exceeds the equilibrium micelle aggregation number in monomer equivalents (i.e. when n > Nagg,mon eq.), the micelle size increases. Beyond this point intra-chain association occurs and the structure is best described as a unimer micelle, in which the average number of polymer chains per micelle is unity (see Table 1). The alkyl chain length still provides an effective packing constraint in the radial direction, so the micelles form increasingly elongated, rod-like structures. Thus it is packing constraints that cause the growth in the micelle rather than the effect of the polymer backbone conformation.
Viscoelastic behaviour with salicylate counterions
Addition of sodium salicylate (NaSal) to both monomeric and micelle-polymerized MUTAB samples produces long, worm-like micelles.19 This effect of certain aromatic anions is well known for conventional cationic surfactant systems such as cetylpyridinium chloride, where strong binding of the salicylate produces rod like micelles and a viscoelastic solution.37,38 In light of the results above, we interpret this as a shift in the threshold for unimer micelle formation in the presence of salicylate to much higher n.In polyMUTAB this transformation is accompanied by a dramatic increase in solution viscosity, although micelles of monomeric MUTAB are little affected. This can clearly be seen in Fig. 5, which shows the steady shear viscosity for 1.5 wt% solutions of MUTABn:NaSal (55:45) at various degrees of polymerization. All samples are shear thinning, with the limiting low-shear viscosity increasing with degree of polymerization. Relaxation times, taken as the inverse of the shear rate at which the viscosity falls to half its initial value, also increase markedly with n. These are listed in Table 2.
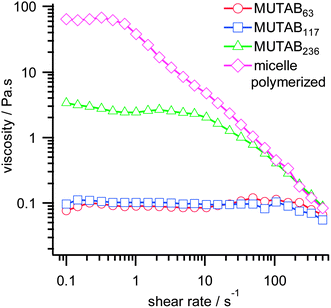 | ||
| Fig. 5 Steady shear viscosity of 1.5 wt% MUTABn:NaSal (55:45 mole ratio) samples in D2O. | ||
| Degree of polymerization, n | τ s/s (steady shear) | τ/s (crossover frequency) | G 0/Pa | d/Å |
|---|---|---|---|---|
| 63 | — | 0.016 | 2.6 | 1170 |
| 117 | 0.0019 | 0.019 | 2.9 | 1120 |
| 236 | 0.04 | 0.27 | 2.8 | 1140 |
| 275 | 0.8 | 2.3 | 6.2 | 872 |
Storage and loss moduli obtained from oscillatory shear measurements are shown in Fig. 6, and are similar to those reported previously for entangled networks of worm-like micelles.37,39 [It was not possible to measure oscillatory rheology on low n samples, or the salicylate-free samples, because of their low viscosity.] Oscillatory data were fit using the Maxwell model40 to extract the plateau modulus and terminal relaxation time, G0 and τ, also listed in Table 2. Although the fits are not particularly good in Fig. 6c and d, the terminal relaxation time, τ, can still be unambiguously determined from the crossover frequency (G′ = G′′). As observed under steady shear, the relaxation time increases with degree of polymerization up to and including the micelle polymerized system (n = 275). Relaxation times in these systems are not more than a few seconds even for the largest degree of polymerization.
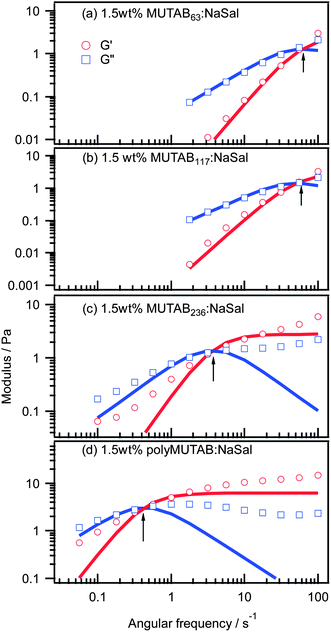 | ||
| Fig. 6 Oscillatory rheology on 1.5 wt% MUTABn:NaSal (55:45 mole ratio) samples in D2O. Arrows indicate crossover frequency. | ||
Relaxation in such living polymer systems occurs by a combination of reptation and chain scission.41 If a system is reptation dominated (i.e. τrep ≪ τbreak) then it can be treated as a classical polymer which, for worm-like micelles yields an exponential relaxation spectrum.41 If chain scission is fast (i.e. τrep > τbreak) then two regions are obtained for the oscillatory rheology data: t ≥ τbreak (low frequency) yields Maxwellian behaviour with a terminal relaxation time τ ∼ (τrepτbreak)1/2, and t < τbreak (high frequency) which is dominated by Rouse motion and produces a minimum in G′′.
At low degrees of polymerization, i.e. MUTAB63:NaSal and MUTAB117:NaSal, both G′ and G′′ are well-described by the Maxwell model in the accessible frequency range. This indicates single relaxation times and stress relaxation dominated by micelle breaking. For longer n, i.e. MUTAB236:NaSal and the MUTABmic:NaSal systems, the terminal relaxation times increase by two orders of magnitude, and the Maxwell model fails to describe G′ and G′′ even at low frequencies. This indicates a transition to reptation dominated stress relaxation and the emergence of a relaxation time spectrum.
The plateau modulus, G0, changes little with degree of polymerization (Table 2). This is not surprising for an entangled network of long, worm-like micelles, for which G0 = kT/d3, where d is the distance between entanglement points.42,43 Here d is approximately 1100 Å for all measured RAFT-MUTABn:NaSal polymer systems, and 870 Å for the micelle polymerized MUTABmic:NaSal system. This also suggests that the observed change in low-shear viscosity is a direct consequence of increasing relaxation time.
As seen from the structures of MUTAB micelles, above a degree of polymerization around 30–60, individual polyMUTA+ chains undergo intrachain association into short rods or prolate spheroidal unimer micelles. Previous SANS studies, together with the observation of entanglement distances, d, much larger than the dimension of any polyMUTAB chains or unimer micelles confirms that addition of salicylate leads once again to multiple chains associating into long, worm-like micelles. By increasing the degree of polymerization of polyMUTA+ chains, the number of potential scission points within each micelle is decreased. This increases τbreak, and gives rise to a transition from scission- to reptation-dominated stress relaxation, beyond which the micelles behave like conventional polymers.
Conclusions
We have synthesised a series of polysoaps to different degrees of polymerization using RAFT controlled polymerization of the T-type cationic MUTAB surfmer and compared their self-assembly structure and dynamics with an in situ micelle polymerized sample.The assembly of these polysoaps remains subject to traditional surfactant packing constraints. At low degrees of polymerization, interchain association of several MUTABn polysoap molecules in aqueous solution form micelles with an approximately constant number of MUTAB monomer equivalents. However, when the degree of polymerization reaches and ultimately exceeds that aggregation number, the assembly transitions into larger unimer micelles. These contain a single polyMUTAB chain formed by intrachain association of its alkyl tails packed into an elongated spheroid or short, rod-like structure with a cross-sectional radius determined by the MUTA+ alkyl chain length.
In the presence of salicylate, these chains assemble into long, worm-like micelles whose dynamics depend strongly on the degree of polymerization of the polysoap. This leads to a transition from scission- to reptation-dominated stress relaxation with increasing degree of polymerization, allowing their terminal relaxation time and zero-shear viscosity to be varied by over two orders of magnitude.
Acknowledgements
We gratefully acknowledge the Australian Institute of Nuclear Science and Engineering (AINSE) for travel funding, Dr Suraj Sharma and Dr Shao Cong Dai for assistance with the rheology measurements, and Professor Sebastien Perrier and Dr Khwanrat Chatjaroenporn for assistance with the RAFT polymerizations. We acknowledge the support of the National Institute of Standards and Technology, U.S. Department of Commerce, and the Korea Atomic Energy Research Institute (KAERI) in providing the neutron research facilities used in this work. This work utilized facilities supported in part by the National Science Foundation under Agreement no. DMR-0944772. This work was supported by Discovery Grants from the Australian Research Council.References
- A. J. Hyde and D. J. M. Robb, J. Phys. Chem., 1963, 67, 2089–2092 CrossRef CAS.
- C. E. Larrabee and E. D. Sprague, J. Polym. Sci., Polym. Lett. Ed., 1979, 17, 749–751 CrossRef CAS.
- S. M. Hamid and D. C. Sherrington, Br. Polym. J., 1984, 16, 39–45 CrossRef CAS.
- C. M. Paleos, Chem. Soc. Rev., 1985, 14, 45–67 RSC.
- S. Hamid and D. Sherrington, J. Chem. Soc., Chem. Commun., 1986, 936–938 RSC.
- P. Dais, C. M. Paleos, G. Nika and A. Malliaris, Makromol. Chem., 1993, 194, 445–450 CrossRef CAS.
- D. Cochin, F. Candau and R. Zana, Macromolecules, 1993, 26, 5755–5764 CrossRef CAS.
- D. Cochin, R. Zana and F. Candau, Macromolecules, 1993, 26, 5765–5771 CrossRef CAS.
- S. R. Kline, Langmuir, 1999, 15, 2726–2732 CrossRef CAS.
- S. Kline, J. Appl. Crystallogr., 2000, 33, 618–622 CrossRef CAS.
- M. Abe, K. Tsubone, T. Koike, K. Tsuchiya, T. Ohkubo and H. Sakai, Langmuir, 2006, 22, 8293–8297 CrossRef CAS.
- S. Liu, Y. I. Gonzalez, D. Danino and E. W. Kaler, Macromolecules, 2005, 38, 2482–2491 CrossRef CAS.
- Z. Y. Zhu, Y. I. Gonzalez, H. X. Xu, E. W. Kaler and S. Y. Liu, Langmuir, 2006, 22, 949–955 CrossRef CAS.
- P. A. FitzGerald and G. G. Warr, Adv. Colloid Interface Sci., 2012, 179, 14–21 CrossRef.
- M. Summers and J. Eastoe, Adv. Colloid Interface Sci., 2003, 100–102, 137–152 CrossRef CAS.
- H.-P. Hentze and E. W. Kaler, Curr. Opin. Colloid Interface Sci., 2003, 8, 164–178 CrossRef CAS.
- K. Chatjaroenporn, R. W. Baker, P. A. FitzGerald and G. G. Warr, J. Colloid Interface Sci., 2009, 336, 449–454 CrossRef CAS.
- K. Chatjaroenporn, R. W. Baker, P. A. FitzGerald and G. G. Warr, Langmuir, 2010, 26, 11715–11719 CrossRef CAS.
- P. A. FitzGerald, K. Chatjaroenporn, X. L. Zhang and G. G. Warr, Langmuir, 2011, 27, 11852–11859 CrossRef CAS.
- J. Michas, C. M. Paleos and P. Dais, Liq. Cryst., 1989, 5, 1737–1745 CrossRef CAS.
- T. P. Le, G. Moad, E. Rizzardo and S. H. Thang, WO9801478A1, 1998.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- C. J. Glinka, J. G. Barker, B. Hammouda, S. Krueger, J. J. Moyer and W. J. Orts, J. Appl. Crystallogr., 1998, 31, 430–445 CrossRef CAS.
- S. R. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- O. Glatter, J. Appl. Crystallogr., 1977, 10, 415–421 CrossRef.
- J. P. Cotton, in Neutron, X-ray and Light Scattering, ed. P. Lindner and T. Zemb, Elsevier Science Publishers, Amsterdam, 1991, p. 11 Search PubMed.
- P. Lindner and T. Zemb, Neutrons, X-rays and light: scattering methods applied to soft condenses matter, Elsevier Science, Amsterdam, 2002 Search PubMed.
- A. Munter, http://www.ncnr.nist.gov/resources/sldcalc.html, accessed 30 October 2012.
- V. F. Sears, Neutron News, 1992, 3, 26–37 CrossRef.
- J. S. Pedersen and P. Schurtenberger, Macromolecules, 1996, 29, 7602–7612 CrossRef CAS.
- W. R. Chen, P. D. Butler and L. J. Magid, Langmuir, 2006, 22, 6539–6548 CrossRef CAS.
- P.-G. de Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, London, 1979 Search PubMed.
- R. Kirste and O. Kratky, Z. Phys. Chem., 1962, 31, 363–374 CrossRef CAS.
- Structure Analysis by Small-angle X-ray and Neutron Scattering, ed. L. A. Feigin and D. I. Svergun, Plenum Press, New York, 1987 Search PubMed.
- J.-P. Hansen and J. B. Hayter, Mol. Phys., 1982, 46, 651–656 CrossRef CAS.
- J. B. Hayter and J. Penfold, Mol. Phys., 1981, 42, 109–118 CrossRef CAS.
- H. Rehage and H. Hoffmann, J. Phys. Chem., 1988, 92, 4712–4719 CrossRef CAS.
- M. E. Cates and S. J. Candau, J. Phys.: Condens. Matter, 1990, 2, 6869–6892 CrossRef CAS.
- A. Khatory, F. Lequeux, F. Kern and S. J. Candau, Langmuir, 1993, 9, 1456–1464 CrossRef CAS.
- J. D. Ferry, Viscoelastic Properties of Polymers, Wiley, New York, 1961 Search PubMed.
- M. E. Cates, Macromolecules, 1987, 20, 2289–2296 CrossRef CAS.
- M. Doi and S. F. Edwards, The Theory of Polymer Dynamics, Clarendon Press, Oxford, 1986 Search PubMed.
- F. Kern, F. Lequeux, R. Zana and S. J. Candau, Langmuir, 1994, 10, 1714–1723 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sm27573f |
| This journal is © The Royal Society of Chemistry 2013 |