 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glycopolypeptide conformations in bioactive block copolymer assemblies influence their nanoscale morphology†
Jessica R.
Kramer‡
a,
April R.
Rodriguez‡
b,
Uh-Joo
Choe
b,
Daniel T.
Kamei
b and
Timothy J.
Deming
*ab
aDepartment of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, USA
bDepartment of Bioengineering, University of California, Los Angeles, CA 90095, USA. E-mail: demingt@seas.ucla.edu; Fax: +1 310-794-5956
First published on 11th February 2013
Abstract
We describe the preparation and assembly of glycosylated amphiphilic diblock copolypeptides, where the hydrophilic glycosylated segments adopt either α-helical or disordered conformations. In this study, glycosylated amphiphilic diblock copolypeptides were prepared using poly(L-leucine), poly(L), as the hydrophobic segment, and poly(α-D-galactopyranosyl-L-lysine), poly(α-gal-K), or poly(α-D-galactopyranosyl-L-cysteine sulfone), poly(α-gal-CO2), as the hydrophilic segment. The poly(α-gal-K) and poly(α-gal-CO2) segments are known to be fully α-helical (>90% at 20 °C) and fully disordered in water, respectively. We found that block copolypeptides containing galactosylated hydrophilic segments of either α-helical or disordered conformation give different assembly morphologies, where the disordered glycopolypeptide segments favor vesicle formation and also present sugar residues that can bind to biological targets.
Introduction
There has been considerable recent interest in the development of multifunctional, nanoscale carriers for targeted delivery of therapeutics.1–3 To precisely control the nanostructured morphology of these delivery vehicles, including shape (e.g. spherical micelles, cylindrical micelles, discs, or vesicles) and internal structure (e.g. spotted, segmented, or core–shell), the assembly of amphiphilic block copolymers in aqueous solution has been highly useful.4,5 However, the incorporation of multifunctionality (e.g. for cellular targeting, uptake, or intracellular release of cargos),6–8 can often perturb the self-assembly process, leading to different morphologies with altered stabilities. We are pursuing the development of block copolypeptide based drug carriers since they are resorbable materials possessing the ordered chain conformations of proteins, which provide an additional means to direct nanostructure independent of many other parameters, such as amino acid composition.9–11 Here, we describe the preparation and assembly of glycosylated amphiphilic diblock copolypeptides, where the hydrophilic glycosylated segments adopt either α-helical or disordered chain conformations. These distinct glycopolypeptide conformations were found to significantly impact block copolymer self-assembly. These results show how careful choice of polypeptide components can be used to direct assembly of nanocarriers into desired morphologies and simultaneously introduce bioactive functionality.Glycosylation of polymeric drug and gene carriers has been shown to lower cytotoxicity, enhance aqueous solubility, and provide targeting to specific cells and organs.12,13 It is also well known in biology that the way in which sugar functional groups are presented greatly affects their ability to bind targets and signal cells.14 We recently reported the preparation of fully glycosylated, high molar mass synthetic polypeptides, which are water soluble and mimic the structures of naturally occurring glycoproteins.15,16 A key feature of these glycopolymers is that their chain conformations are readily controlled, either by choice of peptide backbone or by selective oxidation of side-chain functional groups, such that fully α-helical or fully disordered chains can be obtained. We have now incorporated these glycopolypeptides as hydrophilic segments in amphiphilic diblock copolypeptides to study their aqueous self-assembly and evaluate the properties of the resulting nanostructures. While much is known about how different chain conformations of hydrophobic polypeptide segments influence nanoscale morphology, little is known about the corresponding role played by hydrophilic polypeptide conformations in self assembly.17–20 This has been difficult to study since hydrophilic polypeptide segments presenting similar functionality but differing only in conformation are rare. Here, we have found that block copolypeptides containing galactosylated hydrophilic segments of either α-helical or disordered conformation give different assembly morphologies, where the disordered glycopolypeptide segments favor vesicle formation and present sugar residues that can bind to biological targets (Fig. 1).
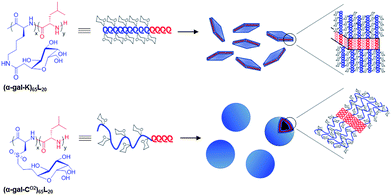 | ||
| Fig. 1 Schematic showing structures of amphiphilic glycosylated diblock copolypeptides and observed self-assemblies. | ||
Previously, we and others reported that amphiphilic diblock copolypeptides containing α-helical hydrophobic segments assemble in water to form spherical, unilamellar vesicles ranging in diameter from tens of nanometers to tens of microns.18,19,21–25 The rod-like conformations of these hydrophobic segments were found to favor side-by-side packing resulting in lamellar vesicle membranes, while samples with disordered hydrophobic segments were found to pack into spherical micelles,26,27 similar to other, conformationally disordered, synthetic block copolymers.5 Block copolypeptides containing hydrophilic segments with either disordered or α-helical conformations in combination with α-helical hydrophobic segments have been found to give vesicular assemblies under certain conditions.18,19,21–25 However, it has been difficult to unequivocally determine the role of the hydrophilic chain conformation in directing nanostructure, since there have been significant differences between the disordered and α-helical hydrophilic segments in these materials (e.g. ionic vs. nonionic). For example, both nonionic poly(Nε-2-(2-(2-methoxyethoxy)ethoxy)acetyl-L-lysine)100-b-poly(L-leucine)20 (KP100L20) with an α-helical hydrophilic segment and ionic poly(L-homoarginine·HCl)60-block-poly(L-leucine)20 (RH60L20) with a disordered hydrophilic segment can be assembled into micron sized vesicles in water.18,19 Some ionic vesicles can be neutralized by adjustment of pH, which results in a transition from disordered to α-helical conformation, but the uncharged, α-helical polypeptide segments are sparingly soluble in water and precipitate above micromolar concentrations.28,29 Likewise, KP100L20 samples with nonionic, disordered hydrophilic segments have been prepared using racemic KP residues, yet inter-chain H-bonding interactions in the resulting disordered segments limit their aqueous solubility as well.18 Consequently, there remains a need for fully α-helical and disordered hydrophilic polypeptide segments that display similar functionality to determine how hydrophilic chain conformation can be used to direct nanoscale assembly and present polypeptide functionality.
Materials and methods
Unless stated otherwise, reactions were conducted in oven-dried glassware under an atmosphere of dinitrogen using anhydrous solvents. Hexanes, THF, DCM, and DMF were purified by first purging with dry nitrogen, followed by passage through columns of activated alumina. Deionized water (18 MΩ cm) was obtaining by passing in-house deionized water through a Millipore Milli-Q Biocel A10 purification unit. All commercially obtained reagents were used as received without further purification unless otherwise stated. Reaction temperatures were controlled using an IKA temperature modulator, and unless stated otherwise, reactions were performed at room temperature (RT, approximately 20 °C). Thin-layer chromatography (TLC) was conducted with EMD gel 60 F254 precoated plates (0.25 mm) and visualized using a combination of UV, anisaldehyde, and phosphomolybdic acid staining. Selecto silica gel 60 (particle size 0.032–0.063 mm) was used for flash column chromatography. 1H NMR spectra were recorded on Bruker spectrometers (at 500 MHz) and are reported relative to residual protons in the deuterated solvents. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity, coupling constant (Hz) and integration. Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet and br, broad. 13C NMR spectra were recorded on Bruker Spectrometers (at 125 MHz). Data for 13C NMR spectra are reported in terms of chemical shift. High-resolution mass spectrometry (HRMS) was performed on a Micromass Quatro-LC Electrospray spectrometer with a pump rate of 20 μL min−1 using electrospray ionization (ESI). All Fourier Transform Infrared (FTIR) samples were prepared as thin films on NaCl plates, and spectra were recorded on a Perkin Elmer RX1 FTIR spectrometer and are reported in terms of frequency of absorption (cm−1). Tandem gel permeation chromatography/light scattering (GPC/LS) was performed on an SSI Accuflow Series III liquid chromatograph pump equipped with a Wyatt DAWN EOS light scattering (LS) and Optilab rEX refractive index (RI) detectors. Separations were achieved using 105, 104, and 103 Å Phenomenex Phenogel 5 μm columns using 0.10 M LiBr in DMF as the eluent at 60 °C. All GPC/LS samples were prepared at concentrations of 5 mg mL−1. The preparation of 2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl-L-lysine-N-carboxyanhydride (α-gal-K NCA),15 2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl-L-cysteine-N-carboxyanhydride (α-gal-C NCA),16L-leucine N-carboxyanhydride (Leu NCA),30 and (PMe3)4Co31 have been previously reported.Preparation of glycosylated diblock copolypeptides
All polymerization reactions were performed in a dinitrogen filled glove box. To a solution of α-gal-K NCA or α-gal-C NCA (1 equiv.) in THF (50 mg mL−1) was rapidly added, via syringe, a solution of (PMe3)4 Co in THF (0.05 equiv., 30 mg mL−1). The reaction was stirred at RT and polymerization progress was monitored by FTIR. Polymerization reactions were generally complete within 3 hours. Immediately upon polymerization completion, aliquots were removed for GPC/LS and endgroup analysis using isocyanate terminated PEG (Mw = 1000 Da).16,32 A solution of Leu NCA in THF (0.33 equiv., 50 mg mL−1) was added and the polymerization was monitored by FTIR. Polymerization reactions were generally complete within 3 hours. After complete consumption of NCA, reactions were removed from the drybox and precipitated into hexanes. Solids were collected by centrifugation and washed with 2 portions of water at pH 2 (HCl), followed by DI water. The polymers were lyophilized to yield white solids (95–99% yield).Molecular weight determination
The degree of polymerization (DP) of the first segment, poly(α-gal-C) or poly(α-gal-K), was determined by 1H NMR end-group analysis of the aliquot end-capped with PEG.32 Integrations were calibrated using the polyethylene glycol chemical shift found at δ 3.64, and the polypeptide DPs were found to be (α-gal-C)65 and (α-gal-K)67. Polydispersity indices were determined using GPC/LS: Mw/Mn for (α-gal-C)65 = 1.09 and Mw/Mn for (α-gal-K)67 = 1.07. The DP of the oligoleucine segments were determined by 1H NMR integrations relative to the glycosylated segment. Final copolypeptide compositions were determined to be (α-gal-C)65L22 and (α-gal-K)67L23.Removal of protecting groups from glycosylated diblock copolypeptides
To a solution of the protected diblock copolymer in DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol 1:2 (10 mg mL−1) was added hydrazine monohydrate (4 equiv./OAc group). The reactions were stirred overnight at room temperature and the product was then observed as a white precipitate. Reactions were quenched by addition of drops of acetone. Et2O was added and the solids collected by centrifugation (99% yield). The solids were taken up with water and transferred to 2000 molecular weight cutoff (MWCO) dialysis tubing and dialyzed against Millipore water for 3 days, with water changes twice per day. Dialyzed copolymers were lyophilized to dryness to yield white fluffy solids (80% yield after dialysis).
:methanol 1:2 (10 mg mL−1) was added hydrazine monohydrate (4 equiv./OAc group). The reactions were stirred overnight at room temperature and the product was then observed as a white precipitate. Reactions were quenched by addition of drops of acetone. Et2O was added and the solids collected by centrifugation (99% yield). The solids were taken up with water and transferred to 2000 molecular weight cutoff (MWCO) dialysis tubing and dialyzed against Millipore water for 3 days, with water changes twice per day. Dialyzed copolymers were lyophilized to dryness to yield white fluffy solids (80% yield after dialysis).
Oxidation of (α-gal-C)65L20
(α-Gal-C)65L20 was dissolved in a mixture of 5% acetic acid and 10% H2O2 in DI water (20 mg mL−1), and the reaction was heated to 38 °C for 16 hours. A few drops of 1 M sodium thiosulfate were added, and then the reaction was transferred to 2000 MWCO dialysis tubing, and dialyzed against Millipore water for 3 days, with water changes twice per day. Dialyzed copolypeptides were lyophilized to dryness to yield poly(α-D-galactopyranosyl-L-cysteine sulfone)65-b-(leucine)20, (α-gal-CO2)65L20, as a white fluffy solid (80% yield after dialysis). 1H NMR (500 MHz, d-TFA, 25 °C): δ 4.75 (s, 65H), 4.49 (s, 155H), 4.36–4.06 (m, 242H), 3.49 (s, 133H), 2.39–1.94 (m, 268H), 1.89–1.64 (m, 132H), 1.35 (s, 24H), 1.08–0.92 (m, 153H).Circular dichroism spectra of diblock glycopolypeptides
Circular dichroism spectra were recorded on an OLIS RSM CD spectrophotometer running in conventional scanning mode. Spectra (190–250 nm) were recorded in a quartz cuvette of 0.1 cm path length with samples prepared using Millipore deionized water. All spectra were recorded as an average of 3 scans. The spectra are reported in units of molar ellipticity [θ] (deg cm2 dmol−1). The formula used for calculating molar ellipticity, [θ], was [θ] = (θ × 100 × Mw)/(c × l) where θ is the experimental ellipticity in millidegrees, Mw is the average molecular weight of a residue in g mol−1, c is the polypeptide concentration in mg mL−1, and l is the cuvette pathlength in cm. The percent α-helical content of the glycopolypeptides was estimated using the formula % α-helix = 100 × (−[θ]222 + 3000)/39000), where [θ]222 is the measured molar ellipticity at 222 nm.33 The measured α-helicity of (α-gal-K)65L20 was 94%.Self assembly of diblock glycopolypeptides in water
Solid (α-gal-CO2)65L20 or (α-gal-K)65L20 was dispersed in THF to produce a 1% (w/v) suspension. The suspension was placed in a bath sonicator for 30 minutes to evenly disperse the polypeptide and reduce large particulates. An equivalent amount of Millipore water was then added to give a 0.5% (w/v) suspension. The suspension became clear as the solution was mixed by vortexing. The mixture was then dialyzed (2000 MWCO membrane) against Millipore water overnight with 3 water changes. Vesicular assemblies were also obtained via slow evaporation of the THF from the mixed solvent suspension.Differential interference contrast (DIC) microscopy
Self assembled copolypeptide suspensions of (α-gal-CO2)65L20 or (α-gal-K)65L20 (0.5% (w/v)) in water were visualized on glass slides with a spacer between the slide and the coverslip (double-sided tape or Secure Seal Imaging Spacer, Grace Bio-labs) allowing the structures to be minimally disturbed during focusing. The samples were imaged using a Zeiss Axiovert 200 DIC/Fluorescence Inverted Optical Microscope.Extrusion of vesicle assemblies
A 0.2% (w/v) aqueous (α-gal-CO2)65L20 vesicle suspension was extruded using an Avanti Mini-Extruder. Serial extrusion of vesicle suspensions were performed through Whatman Nuceleopore Track-Etched polycarbonate (PC) membranes with decreasing filter pore sizes: 3 times through a 1.0 μm filter, 3 times through a 0.4 μm filter, and 3 times through a 0.2 μm filter. The PC membranes and filter supports were soaked in Millipore water for 10 minutes prior to extrusion.Dynamic light scattering (DLS) on extruded vesicles
A 0.2% (w/v) aqueous solution of extruded (α-gal-CO2)65L20 vesicles was placed in a disposable cuvette and analyzed with a Malvern Zetasizer Nano ZS model Zen 3600 (Malvern Instruments Inc, Westborough, MA). A total scattering intensity of approximately 1 × 105 cps was targeted.Laser scanning confocal microscopy (LSCM) of fluorescently labeled vesicles
LSCM images of (α-gal-CO2)65L20 aqueous vesicle suspensions were taken on a Leica Inverted TCS-SP1 MP-Inverted Confocal and Multiphoton Microscope equipped with an argon laser (476 and 488 nm blue lines), a diode (DPSS) laser (561 nm yellow-green line), and a helium-neon laser (633 nm far red line). The copolypeptide suspensions (20 μL of 0.2% (w/v) in Millipore water) were incubated with lipophilic DiOC18 fluorescent dye (1 μL of 0.008% (w/v) in DMSO, Molecular Probes) for 2 hours before imaging. Suspensions of the fluorescently labeled copolypeptides (0.2% (w/v)) were visualized on glass slides with a spacer between the slide and the cover slip (Secure Seal Imaging Spacer, Grace Bio-labs) allowing the self-assembled structures to be minimally disturbed during focusing. Imaging of an xy plane with an optical z-slice (700 nm) showed that the assemblies were water filled, unilamellar vesicles.Transmission electron microscopy (TEM) of extruded vesicles
Extruded (α-gal-CO2)65L20 vesicle suspensions were diluted to 0.1% (w/v) with Millipore water. Samples (4 μL) were placed on a 300 mesh Formvar/carbon coated copper grid (Ted Pella) and allowed to remain on the grid for 60 seconds. Filter paper was used to remove the residual sample. One drop of 2% (w/v) uranyl acetate (negative stain) was then placed on the grid for 90 seconds, and subsequently removed by washing with drops of millipore water and removing the excess liquid with filter paper. The grids were allowed to dry before imaging using a JEM 1200-EX (JEOL) transmission electron microscope at 80 kV.Encapsulation of Texas Red labeled dextran in vesicles
Vesicles composed of (α-gal-CO2)65L20 were prepared as described above, except the aqueous phase contained 0.125 mg mL−1 Texas Red labeled dextran (Mn = 3000 Da). Vesicle solutions were dialyzed in 8000 MWCO tubing overnight to remove unencapsulated Texas Red labeled dextran. As a control to test for dye adsorption to vesicles, pre-formed (α-gal-CO2)65L20 vesicles were incubated for 16 hours with a 0.125 mg mL−1 Texas Red labeled dextran solution and then dialyzed in 8000 MWCO tubing overnight. The samples were then imaged using DIC and LSCM microscopy as described above. No fluorescence was observed in the pre-formed vesicles incubated with Texas Red labeled dextran solution.Evaluation of carbohydrate-lectin binding by turbidity
Ricinus communis Agglutinin I (RCA120) was purchased from Vector labs, and Conconavalin A (Con A) was purchased from Sigma-Aldrich. Lectin solutions were prepared at a concentration of 2 mg mL−1 in 10 mM phosphate, 0.15 M NaCl, pH 7.8. Lectin solutions (600 μL) were transferred to cuvettes and baseline measurements were taken. A solution of (α-gal-CO2)65L20 vesicles (140 nm average diameter) was prepared at a concentration of 1 mg mL−1 in DI water, and 60 μL of the solution was added to cuvettes containing either RCA120 or Con A. Final glycopolypeptide concentrations were 3.3 mM. The solutions were gently mixed and absorbance spectra were recorded over time. All measurements were performed in triplicate.Cell culture
The HeLa cell line was maintained in Minimum Essential Medium (MEM) supplemented with 10% FBS, 1 mM sodium pyruvate, 100 units per mL penicillin, and 100 mg mL−1 streptomycin at a pH of 7.4 in a 37 °C humidified atmosphere with 5% CO2 using standard tissue culture protocols.MTS cell proliferation assay
The MTS cell proliferation assay (CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay) was used to quantify any cytotoxic effects of the (α-gal-CO2)65L20 and RH60L20 vesicle suspensions. HeLa cells were seeded at a density of 4 × 104 cells per cm2 on a 96-well plate prior to the experiment. At the start of the experiment, the cell culture medium was aspirated, and the cells were incubated in medium containing the copolypeptide vesicles for 5 h in a 37 °C humidified atmosphere with 5% CO2. The incubation medium was the same as the cell culture medium except for the absence of FBS, penicillin, and streptomycin. Following the 5 h incubation period, the medium containing copolypeptide vesicles was aspirated. Fresh medium containing 20% MTS was then added to the cells. The cells were placed back into a CO2 incubator for 1 h and then the absorbance at 490 nm (A490) was measured with an Infinite F200 plate reader (Tecan Systems Inc., San Jose, CA, USA). The background absorbance was read at 700 nm (A700) and subtracted from A490. The relative survival of the cells at each polypeptide concentration was quantified by taking the ratio of the (A490–A700) values and comparing between the experimental and control cells.Results and discussion
In this study, glycosylated amphiphilic diblock copolypeptides were designed to incorporate poly(α-D-galactopyranosyl-L-lysine), poly(α-gal-K),15 and poly(α-D-galactopyranosyl-L-cysteine sulfone), poly(α-gal-CO2),16 hydrophilic segments, which are known to be fully α-helical (>90% at 20 °C) and fully disordered in water, respectively. The precursor galactosylated amino acid N-carboxyanhydride (α-gal-K NCA and α-gal-C NCA) monomers15,16 were used to prepare diblock copolymers containing galactose bearing hydrophilic segments ca. 65 residues long connected to α-helical hydrophobic oligoleucine segments ca. 20 residues long, i.e. (α-gal-K)65L20 and (α-gal-C)65L20 (Fig. 1). These chain lengths were chosen to encourage assembly into vesicles, which are desirable nanostructures that can encapsulate both hydrophilic and hydrophobic cargos,6–8 and are based on optimized compositions determined for other vesicle forming diblock copolypeptides.19,22–25 Synthesis of these copolypeptides using (PMe3)4Co initiator in THF yielded samples with narrow chain length distributions and desired compositions,31 and removal of protecting groups gave the galactosylated amphiphilic block copolymers (Table 1).15,16 Since poly(α-gal-C) is partially α-helical in water, we oxidized the thioether linkages in these segments to the corresponding sulfones to produce copolypeptides with fully disordered poly(α-gal-CO2) hydrophilic chains (Fig. 1).16 Circular dichroism analysis of the block copolymers confirmed that (α-gal-K)65L20 is predominantly α-helical in water, and that (α-gal-CO2)65L20 is predominantly disordered in water (Fig. 2).| Block copolypeptide | M n | M w/Mna | Found compositionb | Yieldc (%) | Self-assembled structured |
|---|---|---|---|---|---|
| a Hydrophilic segment lengths (number average molecular weight, Mn, for α-gal-K and α-gal-C segments) and polydispersity index (Mw/Mn) determined using gel permeation chromatography and 1H NMR. b Calculated using 1H NMR. c Total isolated yield of diblock glycopolypeptide. d Structures observed visually using optical microscopy (V = vesicle, A = irregular aggregate, P = plate). | |||||
| (α-gal-C)65L20 | 30910 |
1.09 | (α-gal-C)65L22 | 95 | V |
| (α-gal-K)65L20 | 33130 |
1.07 | (α-gal-K)67L23 | 99 | P, A |
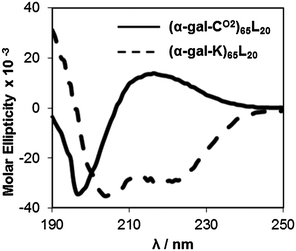 | ||
| Fig. 2 Circular dichroism spectra of glycosylated diblock copolypeptides. Samples are (α-gal-CO2)65L20 (solid line) and (α-gal-K)65L20 (dashed line), 0.2 mg mL−1 in deionized water. Molar ellipticity is reported in millideg cm2 dmol−1. | ||
Attempts were made to assemble vesicles from the galactose containing copolypeptides using mixed solvent annealing, which has been found to assist formation of ordered nanostructures in many other block copolypeptide systems.18,21 The hydrophilic chain conformations of the galactosylated block copolypeptides were found to strongly influence their self assembly in water. The sample with a disordered hydrophilic segment (α-gal-CO2)65L20, gave exclusively vesicles with diameters ranging from hundreds of nanometers to a few microns in diameter (Fig. 3A). The vesicular morphology of the (α-gal-CO2)65L20 assemblies was confirmed by labeling their hydrophobic domains with DiOC18 dye and imaging thin slices through suspensions of the samples using laser scanning confocal microscopy (LSCM), which revealed their membrane structure and hydrophilic interior (Fig. 3C). The ability of these vesicles to encapsulate hydrophilic cargos was also shown by their retention of Texas Red labeled dextran (Mn = 3000 Da) after removal of unencapsulated cargo by dialysis (Fig. 3D). The (α-gal-CO2)65L20 vesicles could also be extruded to obtain low polydispersity nanovesicles with average diameters of 140 nm (PDI = 0.060), which are a desirable size range for use as circulating nanocarriers (Fig. 3E and F).6–8
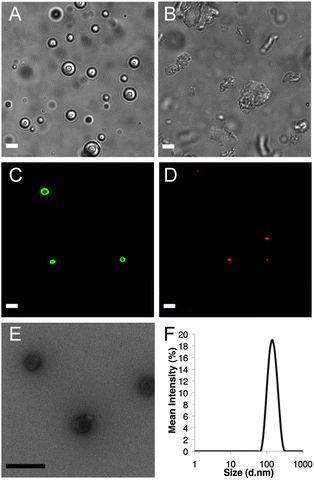 | ||
| Fig. 3 Imaging of glycosylated block copolymer self assemblies. DIC images of (A) (α-gal-CO2)65L20 vesicle suspension and (B) (α-gal-K)65L20 plates and aggregates. LSCM images of (C) (α-gal-CO2)65L20 vesicles containing DiOC18 dye and (D) Texas Red labeled dextran encapsulated within (α-gal-CO2)65L20 vesicles. (E) TEM image of extruded (α-gal-CO2)65L20 vesicles. (F) Size distribution of extruded (α-gal-CO2)65L20 vesicles from DLS. White scale bars = 5 μm, black scale bar = 200 nm. | ||
In contrast to the results above, the highly α-helical sample, (α-gal-K)65L20 gave nearly no vesicles, and instead an abundance of micron sized irregular aggregates and some platelike objects were observed (Fig. 3B). Comparable aggregation behavior has been observed previously in block copolypeptides containing similarly rigid hydrophilic segments.18 The inability of (α-gal-K)65L20 to form vesicles is likely due to rigidity of these hydrophilic domains, which compacts these segments and limits their ability to effectively solubilize and stabilize the assemblies against further aggregation. The rod-like nature of the α-helical hydrophilic segments in (α-gal-K)65L20 also likely acts to stiffen any membranes formed, leading to rigid sheet-like membranes that lack the flexibility needed to accommodate vesicle curvature.18 Related block copolypeptides containing α-helical hydrophilic domains,18 were also found to only form rigid, inflexible membranes regardless of length of the hydrophilic segments used, indicating the helical chain conformation strongly influences assembled morphology.18 The presence of different hydrophilic chain conformations in (α-gal-K)65L20 and (α-gal-CO2)65L20 thus significantly altered their self-assembled structures, where the desired vesicles were favored by the disordered segments in (α-gal-CO2)65L20. The flexibility in the poly(α-gal-CO2) segments also allows better mixing with water, essentially increasing their hydrophilicity, compared to the conformationally rigid poly(α-gal-K) chains. The more open structure of solvated disordered poly(α-gal-CO2) segments should also partially frustrate packing of the rigid hydrophobic oligoleucine segments, making the vesicle membranes themselves more dynamic, flexible and able to accommodate curvature.21
To determine if (α-gal-CO2)65L20 segments retain the bioactivity of their pendant galactose units, we incubated (α-gal-CO2)65L20 vesicles with lectins in a precipitation assay. We chose Ricinus communis agglutinin (RCA120) for the polymer binding lectin since it is known to specifically and selectively bind to galactosyl groups, and concanavalin A (Con A) as a control lectin that binds mannosyl and glucosyl, but not galactosyl, groups.14 When the galactosyl-vesicles were incubated with RCA120, turbidity of the solution was found to increase rapidly from aggregation due to lectin binding (Fig. 4).14,34 Furthermore, the vesicle suspensions showed no increase in turbidity when incubated with Con A, indicating the interaction with RCA120 is a specific binding interaction between this lectin and the galactosyl groups of the polymers.
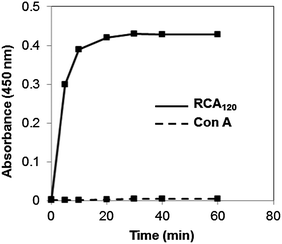 | ||
| Fig. 4 Lectin binding of (α-gal-CO2)65L20 vesicles versus time. Turbidity (absorbance at 450 nm) of (α-gal-CO2)65L20 vesicles (■) when mixed with lectin RCA120 (solid line) or Con A (dashed line), in PBS buffer. Data points are averages of three measurements. All glycopeptide concentrations = 3.3 mM. | ||
Conclusions
Overall, we have found that the chain conformation of hydrophilic polypeptide segments can play a significant role in dictating structure in self assembled nanostructures. The use of hydrophilic segments with disordered conformations in amphiphilic diblock copolypeptides was found to be particularly effective both for formation of vesicular assemblies as well as presentation of functionality in an accessible, active form. The (α-gal-CO2)65L20 vesicles were also found to be minimally cytotoxic (Fig. 5), as compared to cationic polypeptide vesicles such as RH60L20,19 making them attractive for development as biofunctional drug carriers with controlled nanostructure.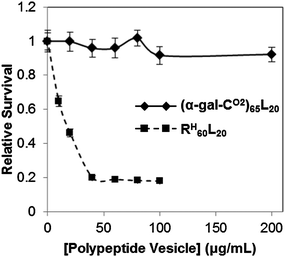 | ||
| Fig. 5 Relative survival of HeLa cells incubated for 5 hours with copolypeptide vesicles determined using the MTS assay. Samples are (α-gal-CO2)65L20 (solid line) and RH60L20 (dashed line). Error bars represent the standard deviation from an average of three measurements. | ||
Acknowledgements
This work was supported by the IUPAC Transnational Call in Polymer Chemistry, the NSF under awards no. MSN 1057970 and no. DMR 0907453, as well as by the Department of Defense Prostate Cancer Research Program under award number W81XWH-09-1-0584.Notes and references
- V. P. Torchilin, Adv. Drug Delivery Rev., 2006, 58, 1532–1555 CrossRef CAS.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640–4643 CrossRef CAS.
- Y. Bae, W.-D. Jang, N. Nishiyama, S. Fukushima and K. Kataoka, Mol. BioSyst., 2005, 1, 242–250 RSC.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2006, 6, 1245–1249 CrossRef CAS.
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2012, 45, 2–19 CrossRef CAS.
- J. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. van Hest, Polym. Chem., 2011, 2, 1449–1462 RSC.
- S. Egli, H. Schlaad, N. Bruns and W. Meier, Polymer, 2011, 3, 252–280 CAS.
- C.-Y. Yang, B. Song, Y. Ao, A. P. Nowak, R. B. Abelowitz, R. A. Korsak, L. A. Havton, T. J. Deming and M. V. Sofroniew, Biomaterials, 2009, 30, 2881–2898 CrossRef CAS.
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS.
- T. J. Deming, Top. Curr. Chem., 2012, 310, 1–171 Search PubMed.
- B. Voit and D. Appelhans, Macromol. Chem. Phys., 2010, 211, 727–735 CrossRef CAS.
- C. Schatz and S. Lecommandoux, Macromol. Rapid Commun., 2010, 31, 1664–1684 CrossRef CAS.
- M. Ambrosi, N. Cameron and B. G. Davis, Org. Biomol. Chem., 2005, 3, 1593–1608 CAS.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2010, 132, 15068–15071 CrossRef CAS.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2012, 134, 4112–4115 CrossRef CAS.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS.
- E. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244–248 CrossRef CAS.
- E. P. Holowka, V. Z. Sun, D. T. Kamei and T. J. Deming, Nat. Mater., 2007, 6, 52–57 CrossRef CAS.
- T. J. Deming, Soft Matter, 2005, 1, 28–35 RSC.
- E. P. Holowka, D. J. Pochan and T. J. Deming, J. Am. Chem. Soc., 2005, 127, 12423–12428 CrossRef CAS.
- J. Rodriguez-Hernández and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef.
- J. Sun, X. Chen, C. Deng, H. Yu, Z. Xie and X. Jing, Langmuir, 2007, 23, 8308–8315 CrossRef CAS.
- E. P. Holowka and T. J. Deming, Macromol. Biosci., 2010, 10, 496–502 CAS.
- A. R. Rodriguez, U.-J. Choe, D. T. Kamei and T. J. Deming, Macromol. Biosci., 2012, 12, 805–811 CrossRef CAS.
- J. A. Hanson, C. B. Chang, S. M. Graves, Z. Li, T. G. Mason and T. J. Deming, Nature, 2008, 455, 85–89 CrossRef CAS.
- J. A. Hanson, Z. Li and T. J. Deming, Macromolecules, 2010, 43, 6268–6269 CrossRef CAS.
- F. Chécot, S. Lecommandoux, Y. Gnanou and H.-A. Klok, Angew. Chem., Int. Ed., 2002, 41, 1340–1343 CrossRef.
- H. Kukula, H. Schlaad, M. Antonietti and S. Förster, J. Am. Chem. Soc., 2002, 124, 1658–1663 CrossRef CAS.
- W. D. Fuller, M. S. Verlander and M. A. Goodman, Biopolymers, 1976, 15, 1869–1871 CrossRef CAS.
- T. J. Deming, Macromolecules, 1999, 32, 4500–4502 CrossRef CAS.
- K. R. Brzezinska, S. A. Curtin and T. J. Deming, Macromolecules, 2002, 35, 2970–2976 CrossRef CAS.
- J. A. Morrow, M. L. Segal, S. Lund-Katz, M. C. Philips, M. Knapp, B. Rupp and K. H. Weigraber, Biochemistry, 2000, 39, 11657–11666 CrossRef CAS.
- J. Huang, C. Bonduelle, J. Thévenot, S. Lecommandoux and A. Heise, J. Am. Chem. Soc., 2012, 134, 119–122 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectral data for all new compounds. See DOI: 10.1039/c3sm27559k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |