 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Patterned chemisorption of proteins by thin polymer film dewetting†
Say Kwang
Lim
,
Sébastien
Perrier
and
Chiara
Neto
*
School of Chemistry, The University of Sydney, NSW 2006, Australia. E-mail: Chiara.neto@sydney.edu.au; Fax: +61-2-9351 3329; Tel: +61-2-93512752
First published on 18th January 2013
Abstract
The covalent attachment of proteins and their patterning on surfaces are important in the bio-medical industry, for use in applications such as bio-sensing and biocompatible medical devices. In this work, proteins are chemisorbed by reaction of their amine groups to glutaraldehyde- and carboxylic acid-functionalised surfaces, within patterns obtained by the dewetting of thin polystyrene films. The proof of principle is shown that consecutive attachment of proteins within the same pattern is possible using this method.
The preparation of surface patterns containing proteins has enabled a number of biomedical applications,1,2 such as the design of biosensors and microarrays, where multiplexing can be used to detect the presence of different types of proteins in a sample mixture.3–5 Protein patterns are used to pattern cells, with the composition and density of extracellular matrix proteins at the surface and the topography of the surface pattern determining cell behaviour and survival.6,7 In these applications, control over the size of the protein-adhesive regions, and the exact conformation, density and type of proteins on the surface are crucial for device function.8–10
We have recently pioneered a method to create micro-scale protein patterns by the dewetting of thin (<100 nm) polymer film bilayers.11–13 Dewetting is a spontaneous process, whereby holes nucleate in an unstable thin liquid film (such as a polymer annealed by exposure to a vapour of a good solvent) on a solid substrate driven by unfavourable intermolecular forces at the interface, the holes grow with time, coalesce with neighbouring holes, and the film is eventually transformed into isolated liquid droplets.14–16 Dewetting by heterogeneous nucleation gives rise to a pattern of uncorrelated holes within the liquid film, which expose the underlying material. The dewetted liquid (polymer) accumulates in a rim around the hole, of height several times greater than the initial film thickness. By stopping the solvent annealing, dewetting can be arrested and a stable pattern of microscopic holes with tuneable size can be created. Thin polymer films are often prepared by spin coating onto planar substrates, but could just as easily be deposited onto irregular three-dimensional objects by alternative techniques such as dip-coating.
Patterning by dewetting is a valid and simple alternative to more sophisticated micro-patterning techniques, such as micro-contact printing, photolithography and microfluidic techniques. This approach has proven to be very effective in producing protein patterns of controlled size and density in a protein-repellent top polymer layer, on top of a protein-adhesive bottom polymer layer. Just recently, we have been able to demonstrate the successful attachment and survival of different cell types within the prepared protein patterns, with potential applications in biomedical devices and single cell studies.12,13 So far we have shown that proteins can be physisorbed within the patterns, but physisorption has some potential drawbacks over chemisorption, such as lack of control on the adsorption process, the potential for protein denaturation upon adsorption, and a relatively short shelf-life.17
This paper for the first time demonstrates the combination of thin film dewetting as a micropatterning approach with the covalent attachment of proteins. Bovine serum albumin (BSA) proteins were chemically attached within the patterns formed by dewetting, using aldehyde- and carboxylic acid-functionalised surfaces. A proof of principle for the successive attachment of proteins on dewetted surfaces is also demonstrated for the first time.
Chemisorbed protein patterns were achieved by the simple steps illustrated in Scheme 1 (Experimental details are presented in the ESI†): bovine serum albumin (BSA) proteins were chemisorbed to a silicon wafer (Si/SiOx) using well-known silanisation and imine bond reactions with glutaraldehyde,18 (parts a, b and c); a polystyrene (PS, 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) thin film was spin-coated onto the glutaraldehyde surface and dewetted via solvent annealing; the dewetted PS film exposed selected regions of the underlying glutaraldehyde (part d); BSA was allowed to chemisorb onto the exposed glutaraldehyde within the dewetted holes (part e, yellow ovals), but also partially physisorbed on the PS (red ovals). The PS film was finally dissolved in toluene, leaving behind a pattern of BSA patches on an unreacted glutaraldehyde surface (part f).
000 g mol−1) thin film was spin-coated onto the glutaraldehyde surface and dewetted via solvent annealing; the dewetted PS film exposed selected regions of the underlying glutaraldehyde (part d); BSA was allowed to chemisorb onto the exposed glutaraldehyde within the dewetted holes (part e, yellow ovals), but also partially physisorbed on the PS (red ovals). The PS film was finally dissolved in toluene, leaving behind a pattern of BSA patches on an unreacted glutaraldehyde surface (part f).
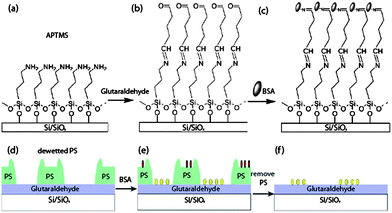 | ||
| Scheme 1 Top row: schematic representation of the chemical functionalization of a silicon wafer with (3-aminopropyltrimethoxy) silane (APTMS) (a), and glutaraldehyde (b) and chemisorption of BSA proteins (c). Bottom row: protein patterning was achieved by dewetting a PS film (d), chemisorption of BSA from solution (e, yellow ovals; red ovals are physisorbed BSA) and dissolution of the PS (f). Graphics not to scale. | ||
The various stages of surface modification of the silicon substrate were characterised by spectroscopic ellipsometry, contact angle goniometry (Table 1), and by Tapping Mode AFM (ESI†). The high value of the measured thickness for APTMS shown in Table 1 compared to the thickness expected for a true monolayer (approximately 0.5 nm)19 attests to the formation of oligomers, which are expected in most silanisation reactions in presence of humidity.20 AFM imaging of an APTMS surface revealed the presence of round aggregates, which are also a common by-product of silanisation in presence of humidity (see Fig. S1 in the ESI†). The presence of amine groups on the APTMS surface was confirmed by ATR-FTIR (Fig. S2†). The relatively high average thickness of the glutaraldehyde layer was also due to self-polymerisation occurring in aqueous solution.21
| Silicon substrate | APTMS | Glutaraldehyde | |
|---|---|---|---|
| a The values in brackets refer to the RMS surface roughness measured including the polymerised aggregates (see Fig. S1).† | |||
| Layer thickness | — | 8 ± 4 nm | 1.3 ± 0.7 nm |
| Advancing contact angle | <5° | 89 ± 3.5° | 86 ± 0.9° |
| Receding contact angle | <5° | 52 ± 6° | 40 ± 6° |
| RMS roughness | 0.2 nm | 0.3 (0.9)a nm | 0.5 (2.3)a nm |
Quartz crystal microbalance (QCM) was used to quantitatively confirm that BSA irreversibly chemisorbed onto the glutaraldehyde surface (Fig. 1, solid line), but only physisorbed onto the APTMS surface (dashed line). An initial increase in adsorbed mass is observed in both traces after the introduction of BSA solution. For the duration of the adsorption process (approximately 30 minutes), the mass adsorbed on the APTMS surface remained constant (∼200 ng cm−2), while increasing amounts of BSA were adsorbed onto the glutaraldehyde surface (up to ∼670 ng cm−2). A sodium dodecyl sulphate solution (SDS, 10%) was flowed over the QCM crystal, a well-known method to remove physisorbed proteins from the surface.22 Initially SDS adsorbed to both surfaces, causing an increase in the adsorbed mass, which was large and constant on the glutaraldehyde surface (∼2450 ng cm−2), while it was smaller and slightly decaying in time (∼470 ng cm−2) over the APTMS surface. This different behaviour suggests that no additional material was adsorbed or desorbed from the glutaraldehyde over time, while BSA proteins were desorbing from the APTMS surface in the same period. After equilibrating in Milli-Q water, the mass on the APTMS surface stabilised at a value slightly below the baseline (∼90 ng cm−2), signifying the removal of all BSA proteins from the surface. The mass remaining on the glutaraldehyde surface was greater than before the SDS washing (∼1910 ng cm−2), which indicates that both BSA and physisorbed SDS surfactant remained. Fluorescence microscopy using a fluorescently labelled BSA (FITC-tagged BSA) confirmed that indeed BSA was still present in large amounts on the glutaraldehyde surface after aggressive SDS washing (Fig. S3, ESI†). The QCM and fluorescence data together confirmed that the BSA was chemically and irreversibly bound to the glutaraldehyde surface.
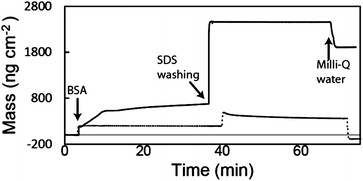 | ||
| Fig. 1 QCM data for the adsorption of BSA on an APTMS surface (dotted line) and on a functionalised glutaraldehyde surface (solid line). | ||
Patterning of the glutaraldehyde surface was achieved by spin-coating a PS film (25 nm thick) and allowing it to dewet via toluene vapour annealing (Fig. 2). The observed dewetting was typical of a liquid film on a non-wettable substrate.12,13,15 During dewetting, holes nucleated in the initially homogenous PS film (part (a)), their diameters grew with time and holes coalesced with neighbouring holes (parts (b and c)), and eventually the system reached its final state, consisting in a number of isolated PS droplets (40–50 μm in diameter) on the glutaraldehyde surface. Remarkably, the size of the holes could be tuned between a few hundred nanometers and a few hundred micrometers, as the dewetting process could be stopped at any moment by removing the substrate from the toluene vapour environment. This simple approach therefore allowed to fix a pattern of holes in the PS film, thus exposing round patches of glutaraldehyde. In this way the amount and location of proteins chemisorbed to the glutaraldehyde surface could be tuned.
 | ||
| Fig. 2 Time lapse optical micrographs of a PS film dewetting from a glutaraldehyde surface. The times shown are from the start of solvent annealing in a toluene vapour environment. | ||
Patterns of PS films at different stages of dewetting were exposed to a solution of FITC-tagged BSA, immersed in an SDS solution (10%, at 70 °C for 1 hour), and then the PS was removed in toluene. Fig. 3(a) shows a fluorescence micrograph of the same surface as in Fig. 2(d) after exposure to FITC-BSA and SDS, and removal of PS: the background glutaraldehyde has a high fluorescence intensity, due to chemisorbed FITC-tagged BSA, while isolated circular regions (40–50 μm in size) present no fluorescence, as in those regions the PS droplets masked the surface and prevented chemisorption of the BSA. The inset in Fig. 3(a) shows a fluorescence image of a similarly prepared glutaraldehyde surface after the protein adsorption process. In this case, the dewetting of the PS film was stopped before completion, at a hole size of approximately 100 μm. As expected, a reverse pattern is observed in this case, with circular regions of high fluorescence intensity surrounded by a background of no fluorescence.
 | ||
| Fig. 3 Fluorescence micrographs of FITC-tagged BSA chemisorbed onto glutaraldehyde surfaces. (a) The areas where the PS droplets masked the glutaraldehyde appear as circular patches with no fluorescence. Inset: a surface similar to that in (a), with dewetted hole size of approximately 100 μm. (b) Same film as in (a) after re-adsorption of FITC-tagged BSA on the previously empty circular patches. Line profiles of the images presented in ESI (Fig. S6†). | ||
The ability to attach proteins to the previously masked glutaraldehyde patches was investigated by re-adsorbing FITC-tagged BSA onto the surface shown in Fig. 3(a). The fluorescence micrograph in Fig. 3(b) displays uniform fluorescence intensity, with the circular regions (with no fluorescence) no longer in evidence, indicating an even coverage of proteins on the surface. This uniform fluorescence confirms that even the areas of the glutaraldehyde layer which were previously coated with PS (and hence non-fluorescent) were still uniformly active and functional. Therefore, by two consecutive steps of dewetting and removal of the PS film, the successive attachment of proteins in different areas of the same substrate was achieved. We are in the process of showing that this step allows to achieve multiplexing, i.e. the co-patterning of more than one type of protein or other biomolecule on the same substrate.
The principle of patterned chemisorption by thin film dewetting is very versatile and could be used with any substrate that is non-wettable by the top polymer film. As a demonstration of this versatility, we investigated chemisorption of proteins and dewetting on a different substrate, a gold surface coated with carboxylic acid functionalities, using another well-known reaction of amine side groups in proteins.23 The carboxylic acid terminal groups in a self-assembled monolayer of 11-mercaptoundecanoic acid on a gold substrate were activated with (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) (EDC) and N-hydroxysuccinimide (NHS) (Scheme shown in Fig. S4†), which then reacted with the amine groups in BSA. The self-assembly process and subsequent activation with EDC and NHS was monitored by QCM (shown in Fig. S5†).
Fig. 4(a) shows QCM data for the adsorption of BSA to the carboxylic acid surface before (dashed) and after activation with EDC and NHS (solid). During the initial BSA adsorption, both profiles showed a similar increase in mass of ∼450 ng cm−2. Upon washing in MES buffer, the adsorbed mass decreased to ∼370 ng cm−2 on the EDC/NHS activated surface and to ∼300 ng cm−2 on the carboxylic acid surface. In both cases, the decrease in mass was due to the removal of physisorbed proteins from the surface. During the washing in SDS surfactant, the adsorbed mass increased to 1350 and 810 ng cm−2 on the EDC/NHS activated and carboxylic acid surfaces, respectively. Similarly to the aldehyde surface in Fig. 1, the adsorbed mass during the SDS washing step on the activated surface remained constant, while a gradual decrease in the adsorbed mass was observed for the carboxylic acid surface. We associate this decrease in mass with the removal of physisorbed proteins from the carboxylic acid surface. After equilibrating again in MES buffer, the adsorbed mass on the carboxylic acid surface was ∼110 ng cm−2, which is approximately 65% lower than the mass adsorbed pre-SDS washing. This small residual mass is likely to include both physisorbed BSA proteins and SDS adsorbed to the surface. In contrast, there was almost no change in the adsorbed mass of BSA on the EDC/NHS functional surface before and after SDS washing (∼350 ng cm−2). This data confirmed that the BSA was chemically and irreversibly bound to the activated EDC/NHS surface.
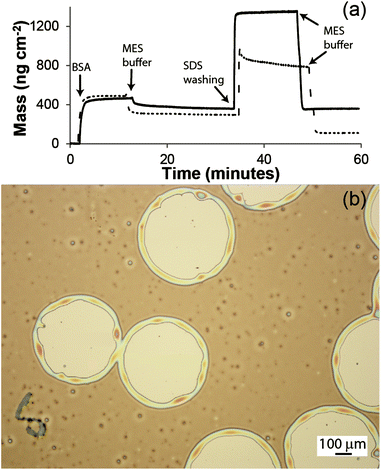 | ||
| Fig. 4 (a) QCM data for the adsorption of BSA on an EDC/NHS functionalised surface (solid line) and a carboxylic acid surface (dotted). (b) Optical micrograph of a PS film dewetted on a carboxylic acid surface. | ||
PS thin films were spun-coated onto the 11-mercaptoundecanoic acid self-assembled monolayer on gold substrate, and dewetting was induced by solvent annealing as in the previous case (Fig. 4(b)). Here the density of nucleated holes appears to be lower than that obtained on the aldehyde surface (and also lower to the typical hole density obtained for similar films on non-wetted substrates12,15,24,25), which leads to a larger maximum hole diameter before coalescence, of the order ∼500 μm. We believe this effect is partially due to an incomplete coating of the gold with the thiol monolayer, and partially due to the surface roughness of the gold substrate (RMS roughness 0.6 nm as measured by AFM). The ability to form holes of larger size is potentially an advantage for applications in the study of single cells, as larger colonies or larger cell sizes could be accommodated. In this approach the patterning of proteins could be achieved by activating only the carboxylic acid groups exposed within the PS holes.
The study of BSA chemisorption and dewetting of PS films enabled us to construct micropatterned protein substrates. This system consisted of dewetted PS films exposing underlying regions of functionalised surfaces able to chemisorb proteins via their amine groups. By removing the PS films after the first chemisorption of BSA, we were able to produce two patterning solutions: either to leave regions with no adsorbed proteins surrounded by regions with chemisorbed proteins, or to backfill the remaining still-functional empty patches on the surface with a second aliquot of proteins. In this latter solution, multiple types of proteins could potentially be made to consecutively chemisorb in different parts of the surface patterns: one protein could be made to attach to a partially dewetted surface, then dewetting could be continued further and a second protein attached on the newly formed rings around the initial holes, and so on. The dewetting could be induced in a non-invasive way, for example by using polymers that can be dewetted by exposure to non-toxic solvents, such as water or dimethyl sulfoxide. In this manner a true example of multiplexing could be achieved, and experiments to demonstrate this potential are underway.
By varying the annealing time, we were able to tune the size of the microscale PS patterns, which in turn dictated the size of the chemisorbed protein domains. Due to the heterogeneous nucleation mechanism, the entire surface was patterned with holes of the same shape and in a narrow size distribution, independent to the overall size of the sample.15 The formation of a laterally ordered pattern of dewetted holes is easily achievable by dewetting, through the use of micro-contact printing.26 However, in most of the applications for which we envisage this approach to be useful, such as the improvement of biocompatibility of medical implants or for single cell culture studies, ordered patterns are not necessary.
This versatile patterning methodology was achieved with simple means, as only basic equipment and commercially available chemicals were required. The patterning step could be performed on large and non-flat substrates by using dip-coating, enabling potential use in biological and biomedical fields. These characteristics of the patterning approach are potentially useful in a range of biomedical applications, such as the development of biosensors and patterned biocompatible coatings for medical implants, and the investigation of the effect of pattern size and distribution on cell behaviour.
Acknowledgements
The authors acknowledge funding from the Australian Research Council. S.K.L. was partly funded by a Gritton scholarship.Notes and references
- M. Dufva and C. B. V. Christensen, Expert Rev. Proteomics, 2005, 2, 41–48 Search PubMed.
- L. Gonzalez-Macia, A. Morrin, M. R. Smyth and A. J. Killard, Analyst, 2010, 135, 845–867 RSC.
- H. Agheli, J. Malmström, E. M. Larsson, M. Textor and D. S. Sutherland, Nano Lett., 2006, 6, 1165–1171 CrossRef CAS.
- G. Singh, S. Pillai, A. Arpanaei and P. Kingshott, Adv. Mater., 2011, 23, 1519–1523 CrossRef CAS.
- M. Kim, J.-C. Choi, H.-R. Jung, J. S. Katz, M.-G. Kim and J. Doh, Langmuir, 2010, 26, 12112–12118 Search PubMed.
- E. Ruoslahti and J. C. Reed, Cell, 1994, 77, 477–478 CrossRef CAS.
- R. G. Flemming, C. J. Murphy, G. A. Abrams, S. L. Goodman and P. F. Nealey, Biomaterials, 1999, 20, 573–588 CrossRef CAS.
- C. J. Wilson, R. E. Clegg, D. I. Leavesley and M. J. Pearcy, Tissue Eng., 2005, 11, 1–18 CrossRef CAS.
- E. Min, K. H. Wong and M. H. Stenzel, Adv. Mater., 2008, 20, 3550–3556 CrossRef CAS.
- Y. Zhang and C. Wang, Adv. Mater., 2007, 19, 913–916 CrossRef CAS.
- C. Neto, Phys. Chem. Chem. Phys., 2007, 9, 149–155 RSC.
- S. C. Thickett, J. Moses, J. R. Gamble and C. Neto, Soft Matter, 2012, 8, 9996–10007 RSC.
- A. M. Telford, L. Meagher, V. Glattauer, T. R. Gengenbach, C. D. Easton and C. Neto, Biomacromolecules, 2012, 13, 2989–2996 Search PubMed.
- G. Reiter, Phys. Rev. Lett., 1992, 68, 75–78 CrossRef CAS.
- R. Seemann, S. Herminghaus, C. Neto, S. Schlagowski, D. Podzimek, R. Konrad, H. Mantz and K. Jacobs, J. Phys.: Condens. Matter, 2005, 17, S267–S290 CrossRef CAS.
- R. Seemann, S. Herminghaus and K. Jacobs, Phys. Rev. Lett., 2001, 86, 5534–5537 CrossRef CAS.
- W. Norde and C. E. Giacomelli, Macromol. Symp., 1999, 145, 125–136 CAS.
- E. Vandenberg, H. Elwing, A. Askendal and I. Lundström, J. Colloid Interface Sci., 1991, 143, 327–335 CrossRef CAS.
- J. H. Moon, J. W. Shin, S. Y. Kim and J. W. Park, Langmuir, 1996, 12, 4621–4624 CrossRef CAS.
- A. Y. Fadeev and T. J. McCarthy, Langmuir, 2000, 16, 7268–7274 CrossRef CAS.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and K. C. Waldron, BioTechniques, 2004, 37, 790 CAS.
- J. L. Bohnert and T. A. Horbett, J. Colloid Interface Sci., 1986, 111, 363–377 Search PubMed.
- J. C. Sheehan and G. P. A. Hess, J. Am. Chem. Soc., 1955, 77, 1067–1068 CrossRef CAS.
- S. C. Thickett, A. Harris and C. Neto, Langmuir, 2010, 26, 15989–15999 CAS.
- A. M. Telford, S. C. Thickett, M. James and C. Neto, Langmuir, 2011, 27, 14207–14217 CrossRef CAS.
- M. Ghezzi, S. C. Thickett and C. Neto, Langmuir, 2012, 28, 10147–10151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; AFM, ATR-FTIR, fluorescence and QCM data; scheme of carboxylic acid activation. See DOI: 10.1039/c2sm27241a |
| This journal is © The Royal Society of Chemistry 2013 |