 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of glycosaminoglycans on lipid dynamics in supported phospholipid bilayers
Harekrushna
Sahoo
ab and
Petra
Schwille
*ac
aBIOTEC, TU Dresden, Tatzberg 47-51, Dresden, Germany
bNational Institute of Technology, Rourkela, India
cMPI of Biochemistry, Am Klopferspitz 18, 82152 Martinsried, Germany. E-mail: schwille@biochem.mpg.de
First published on 25th February 2013
Abstract
Glycosaminoglycans (GAGs) are important constituents of extracellular matrices (ECMs). As charged polymers, they do most likely influence lipid and protein dynamics in the outer leaflet of plasma membranes. In this study, we investigated their specific effect, depending on concentration, on lipid diffusion in model membranes. In our assay, GAGs are simply attached electrostatically to supported phospholipid (DOPC) bilayers doped with small amounts of cationic lipid (DOTAP) at physiological pH. Lipid dynamics are characterized via the diffusion of fluorescent lipid analogs (DiD/DiO), determined by fluorescence correlation spectroscopy (FCS). We find that diffusion of DiD is significantly affected by the attachment of GAG. Quite surprisingly, short chains (≤10 disaccharide units) of hyaluronic acid (unsulfated GAG) on the membrane surface affect the DiD diffusion coefficients stronger than medium or long chains (≥100 disaccharide units). In particular, short chains of hyaluronic acids at micromolar concentrations display a 2-fold decrease of the diffusion coefficients compared to the situation without GAG. At nanomolar concentrations of hyaluronic acid of both short and long chains, DiD diffusion remains unaltered. In contrast, sulfated GAGs, such as heparan sulfate (HS) and heparin, affect the lipid diffusion already at sub-micromolar concentrations, albeit not as strongly, with a less than 1.5 fold reduction of the diffusion coefficient. Chondroitin sulfate, another class of sulfated GAGs, did not impose any effect on DiD diffusion in the supported phospholipid bilayer at the concentrations studied. We also investigated desulfated heparin, to explore the role of sulfation and to compare its effect with HA. It is observed that heparin derivatives with lower degrees of sulfation have little effect on the lipid diffusion. Altogether, our results suggest that the presence of certain carbohydrate polymers in the ECM does have a noticeable effect on lipid dynamics in biological membranes.
Introduction
Glycosaminoglycans (GAGs), an evolutionary well-conserved class of carbohydrates, are important components of the extracellular matrix (ECM), attached to the outer surface of the cell membrane.1,2 Structurally, GAG, a polymer chain of repeated disaccharide units (acidic; hexouronic acid and amino; hexosamine monosaccharides, Table 1), is covalently linked to a protein to form proteoglycans or glycoproteins, depending on the ratio between GAG and the protein.3 Amino sugars are a class of compounds with diverse and ubiquitous functions, having few counterparts in the biological field.4–6 Members of the GAG family are classified depending on their constituent sugar molecules, i.e., hexosamine (galactosamine and glucosamine), hexose (galactose), and hexuronic acid (glucoronic acid and iduronic acid). They also vary in the geometry of the glycosidic linkage and the substitution of sulfate groups on the sugar molecules. Basically, hyaluronic acid (or hyaluronan: HA) is a non-sulfated GAG, where as chondroitin sulfate (CS), keratin sulfate (KS), heparan sulfate (HS), and heparin are sulfated GAGs. GAGs are highly negatively charged, due to the carboxylic acid and substituted sulfate groups.7 The net negative charges on the GAG molecules attract cations such as Na+, and after binding to sodium ions, they interact with the water molecules.8,9 Some specific functions of GAGs result from their interaction with solvent molecules: heparin as an anti-coagulant, hyaluronan as a component in the synovial fluid lubricant in body joints, and chondroitins, which can be found in connective tissues, cartilage, and tendons.10–12| Types of GAG | Chemical structure | Monosaccharide units | Employed chain length (approx.) | Molecular weights (in this context) |
|---|---|---|---|---|
| a CS and HS can be obtained in different forms. From the sulfation point of view, CS can have the sulfation in glucoronic acid or N-acetyl galactose (N, 4 and 6 positions), where as HS can have the sulfations at the same position as in CS but mostly, HS is obtained as monosulfated disaccharides. | ||||
| Hyaluronic acid (HA) |

|
Glucoronic acid and N-acetyl galactosamine | 10 | 3.63 kDa |
| 240 | 95.0 kDa | |||
| 2330 | 921.0 kDa | |||
| 2530 | 1000 kDa | |||
| Chondroitin sulfate (CS) |

|
Glucoronic acid and N-acetyl galactosamine | 102 | 65.0 kDa |
| Heparan sulfate (HS) |

|
Glucoronic acid and N-acetyl galactosamine | 20 | 12 kDa |
| 28 | 17 kDa | |||
| Heparin |
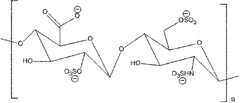
|
Iduronic acid and N-acetyl galactosamine | 23 | 14 kDa |
GAGs, together with other components of the extracellular matrix, are supposed to be involved in many biological functions at and across the cell membrane. In particular, signal transduction, initiated by the interaction between soluble ligands and their receptors, may be strongly influenced by the presence of an extracellular matrix.13 Particularly the glycocalyx, an extracellular layer formed by “glycolipids” upon attachment of carbohydrates to lipids, may provide a diffusion barrier and thus, a certain protection to the cell. The specific role of this outer glycocalyx layer, as well as its counterpart in the gel-like intercellular matrix has so far not been fully elucidated. As one aspect, it is quite plausible that the presence of an ECM significantly affects lipid and protein dynamics in the membrane, which also impacts on essential lipid–protein interactions.
In 2007, Zhang and coworkers reported that the presence of a polymer, quaternized poly(4-vinylpyridine), on the membrane surface creates heterogeneity in the lipid diffusion of the bilayer. They observed a significant difference between the diffusion coefficients of the lipids which are in contact with the polymer (0.50 ± 0.12 μm2 s−1) and those which are not (2.62 ± 0.18 μm2 s−1).14 Since then, little more has been reported on carbohydrate attachment to membranes and their effects on the lipid dynamics. In a recent study,15 the effect of mucin glycoprotein, end-functionalized by hydrophobic anchors to incorporate it into the lipid bilayer, on the lipid dynamics was characterized.16 Basically, the incorporation of derivatized mucins (extended with N-acetyl galactosamine, which is one of the monosaccharides in the GAG disaccharide unit) into the cell surface did not alter lipid mobility in the bilayer17 Furthermore, Quemeneur and coworkers studied the adsorption of HA on a DOPC membrane system as a function of pH.18,19 Although there have been a few recent studies on carbohydrate quantification on membrane surfaces and the effects on the membrane topology, the relationship between the structure and conformation of GAGs and the dynamics of lipid and protein diffusion, and other processes in the bilayer, remains largely unexplored.
Here, we specifically focus on the effect of different types and concentrations of GAGs on lipid diffusion in supported lipid bilayers (SLBs). We used four different GAGs: HA, CS, HS, and heparin (Table 1), with nano- to millimolar concentrations, depending on the solubility of the GAGs. In the case of HA, we also used three different chain lengths, i.e., LHA (low hyaluronic acid), MHA (medium hyaluronic acid), and HHA (high hyaluronic acid). The polymers are attached to the supported lipid bilayer by ionic interactions between negatively charged GAG and positively charged lipid DOTAP. As probes for lipid diffusion, we use the lipid analogues DiD and DiO, their diffusion properties are determined by fluorescence correlation spectroscopy (FCS). FCS has several advantages over FRAP, which is also used to determine diffusion parameters, such as the requirement of lower fluorescent probe concentration (0.005 to 0.01%), higher spatial resolution, and a better precision in determining diffusion heterogeneity.20–23
Material and methods
Materials
Lipids (DOPC: 1,2-dioleoyl-sn-glycero-3-phosphocholine and DOTAP: 1,2-dioleoyl-3-trimethylammonium-propane) were purchased from Avanti Polar Lipids. Lipid fluorescent dyes, DiD (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine) and DiO (3,3′-dioctadecyloxacarbocyanine), were obtained from Invitrogen. Glycosaminoglycans (GAGs), such as heparan sulfate (HS) and chondroitin sulfate (CS), were purchased from Sigma and hyaluronic acid (HA) was obtained from Innovent (Jena, Germany). For the experimental purpose, we used three different classes of HA, which are classified on the basis of their molecular weights (low: 3.6 kDa, medium: 95 kDa and high: 1000 kDa). Tris was purchased from Sigma. For control experiments, fluorescein labeled HA, CS, HS, and heparin were obtained from PG Research (Tokyo, Japan) and labeled protein, Alexa 594 labeled Wheat Germ Agglutinin (WGA), from Invitrogen. Unlabeled heparin and its derivatives were obtained from Prof. Carsten Werner (Max Bergmann Center of Biomaterials, Dresden, Germany).Lipid membranes
Supported lipid bilayers (SLBs) were formed by standard vesicle fusion techniques.24 The bilayers were composed of 95.0 mol% DOPC, a zwitterionic lipid, 5.0 mol% DOTAP, a positively charged lipid, and 0.01 mol% DiD, a fluorescent lipid probe. To rule out a dependence of the measurements on the specific lipid probe, DiO was used as an alternative to DiD. The procedure of SLB formation started with the formation of dry thin lipid films from the mixture of required lipids (DOPC, DOTAP and DiO/DiD) by evaporating the solvent. Then the dry thin lipid film was rehydrated with SLB buffer (10 mM HEPES, 150 mM NaCl and pH 7.4) to obtain MLVs (multi lamellar vesicles). In the following step, the MLVs were sonicated at room temperature to obtain SUVs (small unilamellar vesicles). In the final step, SUVs were burst in the presence of Ca2+ ions and spread onto the mica surface, which was previously glued to the glass surface using UV-adhesive (inert towards fluorescence). The remaining SUVs (which did not burst on the mica surface) were removed by excess washing with the required buffer (Tris 20 mM, pH 7.0).The bilayer was incubated with the glycans for 45 minutes, unbound glycan was washed away, and the resulting membrane was imaged by fluorescence microscopy using a LSM 510 (Zeiss) inverted fluorescence microscope and a photo multiplier tube (PMT).
Fluorescence correlation spectroscopy (FCS)
FCS was employed to measure the lateral lipid mobilities and to assess the impact of glycans on lipid dynamics in supported lipid membranes.20 The experiments were performed on a commercial FCS unit, based on an LSM 510 (Confocor 3, Zeiss) inverted fluorescence microscope. In brief, a 488/633 nm (as per the requirement) beam from Ar-ion/He–Ne laser was coupled into the light path of the microscope through an optical fiber and focused by the water immersion objective (40× magnification, 1.2 NA) onto the sample. A hardware correlator translates the photon arrival pulses into intensity fluctuations and calculates the correlation in real time. The correlation curves were acquired and fitted to analytical expressions (eqn (1)). For an averaged correlation measurement, a minimum of 10 separate correlation measurements of each 10 seconds duration were taken, and corresponding standard deviations for every point of the experimental curves were calculated from multiple experiments.The following analytical form of the temporal auto-correlation G(τ) was used to obtain the diffusion time of a fluorescent molecule through the confocal observation volume in a two-dimensional membrane system:
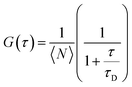 | (1) |
Diffusion coefficients (D) and diffusion coefficient ratio (Dratio)
The diffusion coefficient of the lipids (particularly DiD or DiO in this case) in the supported lipid bilayer was determined from the experimentally obtained correlation time (τD) and the known waist (r0) of the confocal observation volume using the following equation: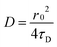 | (2) |
The waist of the observation volume, r0, was determined independently from calibration measurements using 50 nM Alexa 488 and 647 (Invitrogen Corp.) in 50 mM Tris of pH 7 and employing the known diffusion coefficient of the fluorophore (435 μm2 s−1 for Alexa 488 and 330 μm2 s−1 for Alexa 647 in water) as a standard.25,26 The obtained r0 value is used to calculate the unknown diffusion coefficients of lipid fluorescent markers (DiD/DiO) in the lipid bilayer systems in the presence and absence of GAG on its surface.
D ratio is the ratio between the diffusion coefficients of DiD or DiO in the absence (Dwithout GAG) and presence (Dwith GAG) of GAG. Dratio can be written as follows:
| Dratio = Dwithout GAG/Dwith GAG | (3) |
Results and discussions
The effect of different GAGs on lipid diffusion was studied by FCS in terms of Dratio (the ratio of the DiD diffusion coefficient without and with GAG), providing qualitative information about the interaction between the GAG polymer and the membrane, in particular, DOTAP. The use of the dimensionless quantity Dratio eliminates the error in determining exact diffusion coefficients, making it independent of the membrane preparation or the influence of the support. Altogether, larger values for Dratio indicate higher impact of the GAG on lipid diffusion. The diffusion parameters obtained and reported in this article are considered as the averaged result from a heterogeneous membrane system created by the GAG after interacting with lipid bilayer. Along with FCS, fluorescence imaging is used to probe the attachment of fluorescently labeled GAG polymers (Fluorescein-labeled HA, HS, CS, and heparin) with the membrane.Effect of glycosaminoglycan attachment
GAG is a highly negatively charged constituent of the ECM, and in order to attach different types of GAG polymers, positively charged lipid (i.e., DOTAP) was doped into the artificial supported phospholipid bilayers (DOPC: 95%, DOTAP: 5% and DiO or DiD: 0.01%). As a proof of GAG attachment to the membrane surface, fluorescence imaging and FCS measurements were performed on the supported lipid bilayers on a mica surface. As shown in Fig. 1A, the fluorescence intensities were recorded as a function of membrane height (z-stack). The gradual decay over many μm, in spite of the small (few nm) thickness of the membrane, reflects on the optical properties of the detection volume. In Fig. 1A, the blue dashed line represents the fluorescence of DiO (excited with 488 nm) in the membrane. Fluorescence of DiO was followed before (blue square) and after (blue circle) addition of LHA (low HA, Mw: 3.63 kDa) in order to observe the membrane surface homogeneity. To confirm LHA attachment, a sugar binding protein, WGA: Wheat Germ Agglutinin (a class of Lectin proteins), conjugated to Alexa 594 was used as a fluorescent probe, as it selectively binds to the N-acetyl galactosamine of the GAG disaccharide unit. The fluorescence signal of Alexa 594 conjugated WGA (excited with 543 nm) is represented by a red dashed line in Fig. 1A.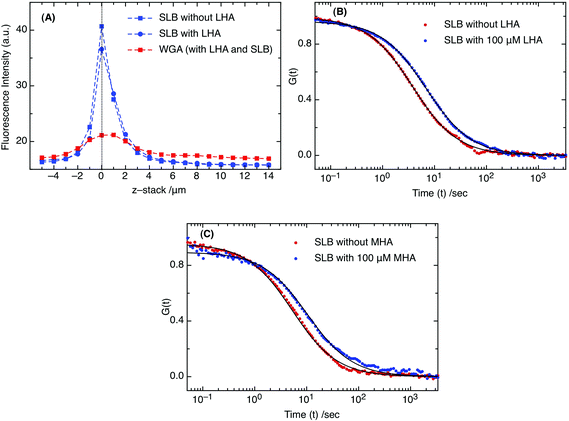 | ||
| Fig. 1 (A) Fluorescence intensities of DiO in SLB without LHA (blue squares) and with LHA (blue circles) and A594-WGA (red squares) when attached to LHA on the SLB surface. (B) Diffusion curves (from FCS measurements) indicating the differences in the presence (red) and absence (green) of 100 μM LHA and (C) 100 μM MHA. The solid lines represent the fitting curves for corresponding FCS curves. | ||
The strong variation in the fluorescence signal of probe WGA as a function of distance from the membrane (z-stack) confirms the attachment of GAG to the membrane surface. Consequently, FCS measurements on DiD (excited at 633 nm) indicate a significant decrease in diffusion coefficient in the presence of LHA (Fig. 1B and C). In a control measurement to exclude a potential role of electrostatic effects between the positively charged DiD/DiO and GAG, SLBs without DOTAP showed no difference in DiD/DiO diffusion coefficients in the presence and absence of LHA. Their inertness towards GAG in contrast to DOTAP could be due to their small sized head groups. A pioneering study by Axelrod in 1979 suggested that cyanine fluorescent markers (i.e., DiI, which is similar to DiD/DiO and carries a positive charge) lie parallel to the phospholipids on the membrane surface.27 DOTAP, on the other hand, has a bulkier head group compared to DiD or DiO and thus, is more solvent accessible.
Taken together, we observe no direct interaction between GAG and DiD/DiO. In contrast, the fluorescence microscopy and FCS measurements in the presence of DOTAP clearly point to significant interactions between the positively charged membrane surface and the negatively charged GAG molecules (Fig. 2A), which in turn affect lipid diffusion. The absolute intensities of the fluorescein-GAG polymers, and thus, the absolute binding affinities, are difficult to compare, as the fluorescein labeling is not specific (0.5 to 0.7 fluorescein molecule per disaccharide unit depending on the GAG type, as specified by the manufacturer).
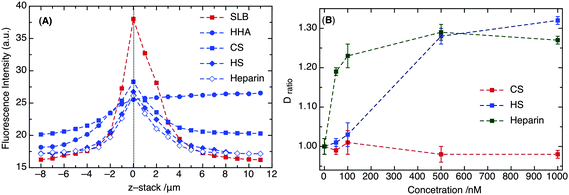 | ||
| Fig. 2 (A) Fluorescence intensities of DiD (red square) and different fluorescein-labeled GAG polymers (in blue). The dotted line at z-stack = 0 indicates the membrane surface. (B) Comparison of Dratio of DiD for different sulfated GAGs (i.e., HS, heparin and CS) at nanomolar concentrations. | ||
Structure-related differences, effects of sulfation
In HA, the carboxylate ion is the only interacting moiety, and in the case of sulfated GAGs, both carboxylate and sulfate groups are likely responsible for membrane binding (structural representation, Table 1). To elucidate potential differences, we compared four different fluorescein labeled GAG molecules, i.e., fluorescein-HHA (Mw: 921 kDa), fluorescein-CS (Mw: 65 kDa), fluorescein-HS (Mw: 17 kDa), and fluorescein-heparin (Mw: 12 kDa). For the latter three, the fluorescence signals of DiD and fluorescein-GAG decrease proportionally when moving below or above the membrane (Fig. 2A), suggesting a strong interaction of GAG with DOTAP. In contrast, the signal of fluorescein-HHA does not show any peak at the membrane surface, but rather stays constant above it, suggesting that it remains largely in solution. In accordance with this observation, there is no noticeable effect on DiD diffusion for HHA, in contrast to the other GAGs. DiD shows an increase in diffusion time (4.0 ms to 10 ms, Fig. 1B) upon attachment of 100 μM LHA, which can be translated to a decrease in diffusion coefficient: from 2.49 × 10−8 cm2 s−1 without to 1.24 × 10−8 cm2 s−1 with. Similarly, for 100 μM MHA, the diffusion coefficient drops from 2.81 × 10−8 cm2 s−1 in the absence to 1.63 × 10−8 cm2 s−1 in the presence (Fig. 1C). The change in DiD diffusion is indicative of the GAG interaction with the DOTAP head groups on the membrane surface.The basic differences among the three sulfated GAGs are their degree of sulfation (number of sulfate groups on one disaccharide unit), chain length, and chemical structure (occurrence of different amino-sugars in disaccharide units). HS and heparin show a noticeable effect on lipid diffusion already at high nanomolar concentrations, whereas CS does not (Fig. 2B). The observed variations in Dratio between CS, HS and heparin could be explained through their differences in chemical structure, including chain length and degree and point of sulfation. The differences in the solution structures of HS and heparin indicate that heparin has 50% trisulfate and 50% disulfate forms, and all the representative disaccharide units are sulfated. In contrast, HS has a repeat of sulfated (either mono- or di-sulfated) and non-sulfated disaccharide units.28 As a result of heavy sulfation in heparin, the concentration dependence of lipid diffusion in the presence of heparin does not seem to display a cooperative effect, compared to HS, where the curve is more sigmoidal (Fig. 2B). In contrast, HS shows a cooperative effect on DiD diffusion upon attachment to the DOTAP in the lipid bilayer. Compared to the structure of heparin, CS has also continuous repeats of sulfated disaccharide units, but it has glucoronic acid instead of iduronic acid like HS.29,30 The conformational change in the acidic sugar of CS might result in a weaker binding with DOTAP. Consequently, there is almost no effect of CS on the DiD diffusion (Fig. 2B).
Additionally, a comparison of fully sulfated heparin with its desulfated derivative (d-heparin) has been carried out to investigate the role of sulfation. The most important aspect about these derivatives is that the chain length remains the same for all and thus, the effect of chain length can be eliminated. Besides the idea of comparing the role of sulfation, the motive of using desulfated heparin is to compare it with LHA, due to their structural similarity. Table 2 shows a decrease in Dratio from heparin to d-heparin, indicating the impact of sulfate groups on the lipid diffusion in the supported lipid bilayer at several concentrations. As a function of concentration (from 50 to 1000 nM), both derivatives show slight variations in the diffusion coefficient values, but the trend is not clear. Although d-heparin has a structural similarity with HA, the impact on lipid diffusion is different. This effect could be due to the conformational differences in the disaccharide units of HA and d-heparin.

Polymer length dependence
Different lengths of HA polymer as the only unsulfated GAG were used to gain insight into the role of chain length on the interaction between GAG and DOTAP. Besides LHA as mentioned above, the lipid diffusion was also observed in the presence of MHA (medium HA, Mw: 95 kDa) and HHA (high HA, Mw: ∼1000 kDa). LHA and MHA have significant impact, as a function of concentration, on the DiD diffusion, as revealed from the experimental observations (shown in Fig. 3). A pronounced effect on diffusion, quantified by Dratio, could first be observed at low micromolar concentrations of LHA and MHA (Fig. 3), further increasing up to 100 μM. In contrast, the diffusion coefficient remained unaffected (within error) in the presence of HHA. At 10 μM concentration, Dratio in the presence of LHA and MHA are 1.61 and 1.19, respectively, whereas it is 0.97 for HHA. This difference could be a result of the conformational change in the HA family as a function of chain length. It is conceivable that the binding site in HHA (i.e., carboxylic acid) is not or partially exposed to the solvent, restricting the interaction with membrane surface, compared to MHA and LHA.31,32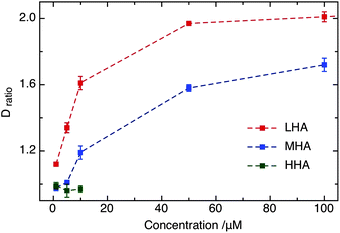 | ||
| Fig. 3 Comparison of Dratio of DiD for different unsulfated GAGs (i.e., LHA, MHA and HHA) at micromolar concentration. | ||
Conclusions
In this study, we investigated the impact of several gylcosaminoglycans (GAGs), being essential constituents of extracellular matrixes, on the dynamics of lipid molecules in membranes. Using electrostatically attached GAGs of negative charge on supported model membranes doped with positively charged lipids (DOTAP) as anchoring sites, we demonstrated that the diffusional mobility of lipids within the GAG-decorated membrane slowed down significantly, depending on concentration and the exact chemical nature of the GAGs. A strong interaction between GAG and membrane is observed in the case of short chains of GAG (i.e. low hyaluronic acid) and sulfated GAG (i.e. heparan sulfate), suggesting the roles of chain length and degree of sulfation. The finding that diffusing molecules within the membrane are not only directly but also indirectly affected by GAG attachment has important consequences when discussing a possible role of extracellular matrixes for lipid and protein dynamics in cell membranes, and thus, essential processes like cellular signaling. For defining appropriate future in vitro model systems of key physiological processes, constituents of the extracellular matrix will have to be taken more carefully into consideration.Acknowledgements
We thank Dr. Carsten Werner for providing the heparin and its derivatives, and for valuable discussions. Emiliya Pogosyan assisted in the data acquisition and reproduction. Financial support by the German research foundation (DFG) within the Transregio SFB 67 is gratefully acknowledged.References
- R. Raman, V. Sasisekharan and R. Sasisekharan, Chem. Biol., 2005, 12, 267–277 CrossRef CAS.
- T. N. Laremore, F. M. Zhang, J. S. Dordick, J. Liu and R. J. Linhardt, Curr. Opin. Chem. Biol., 2009, 13, 633–640 CrossRef CAS.
- N. S. Gandhi and R. L. Mancera, Chem. Biol. Drug Des., 2008, 72, 455–482 CAS.
- M. R. J. Salton, Annu. Rev. Biochem., 1965, 34, 143–174 CrossRef CAS.
- W. Koopmann, C. Ediriwickrema and M. S. Krangel, J. Immunol., 1999, 163, 2120–2127 CAS.
- M. Hook, L. Kjellen, S. Johansson and J. Robinson, Annu. Rev. Biochem., 1984, 53, 847–869 CrossRef CAS.
- R. P. Mecham, The Extracellular Matrix: an Overview, Springer-Verlag, Heidelberg, 2011 Search PubMed.
- M. P. McGee and J. Liang, J. Biol. Chem., 2001, 276, 49275–49282 CrossRef CAS.
- E. C. Stanca-Kaposta, D. P. Gamblin, E. J. Cocinero, J. Frey, R. T. Kroemer, A. J. Fairbanks, B. G. Davis and J. P. Simons, J. Am. Chem. Soc., 2008, 130, 10691–10696 CrossRef CAS.
- A. Varki, R. Cummings, J. Esko, H. Freeze, G. Hart and J. Marth, Essentials of Glycobiology, Cold Spring Harbor Laboratory, California, 1999 Search PubMed.
- V. Prabhakar, I. Capila and R. Sasisekharan, in Glycomics: Methods and Protocols, Humana Press Inc, 999 Riverview Dr, Ste 208, Totowa, NJ 07512-1165 USA, 2009, pp. 331–340 Search PubMed.
- N. Itano, J. Biochem., 2008, 144, 131–137 CrossRef CAS.
- R. J. Linhardt and T. Toida, Acc. Chem. Res., 2004, 37, 431–438 CrossRef CAS.
- L. F. Zhang and S. Granick, Macromolecules, 2007, 40, 1366–1368 CrossRef CAS.
- E. Gemma, O. Meyer, D. Uhrín and A. N. Hulme, Mol. BioSyst., 2008, 4, 481–495 RSC.
- D. Rabuka, M. B. Forstner, J. T. Groves and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 5947–5953 CrossRef CAS.
- K. Godula, M. L. Umbel, D. Rabuka, Z. Botyanszki, C. R. Bertozzi and R. Parthasarathy, J. Am. Chem. Soc., 2009, 131, 10263–10268 CrossRef CAS.
- F. Quemeneur, M. Rinaudo, G. Maret and B. Pépin-Donat, Soft Matter, 2010, 6, 4471–4481 RSC.
- F. Quemeneur, M. Rinaudo and B. Pepin-Donat, Biomacromolecules, 2008, 9, 2237–2243 CrossRef CAS.
- E. Haustein and P. Schwille, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 151–169 CrossRef CAS.
- S. Chiantia, J. Ries and P. Schwille, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 225–233 CrossRef CAS.
- D. Grunwald, M. C. Cardoso, H. Leonhardt and V. Buschmann, Curr. Pharm. Biotechnol., 2005, 6, 381–386 CAS.
- J. Widengren and R. Rigler, Cell. Mol. Biol., 1998, 44, 857–879 CAS.
- S. Chiantia, N. Kahya and P. Schwille, Langmuir, 2005, 21, 6317–6323 CrossRef CAS.
- A. Loman, B. C. Müller, F. Koberling, W. Richtering and J. Enderlein, in 14th International Workshop on Single Molecule Spectroscopy and Ultrasensitive Analysis in Life Sciences, Berlin, Germany, 2008 Search PubMed.
- Z. Petrášek and P. Schwille, Biophys. J., 2008, 94, 1437–1448 CrossRef.
- D. Axelrod, Biophys. J., 1979, 26, 557–574 CrossRef CAS.
- S. Khan, E. Rodriguez, R. Patel, J. Gor, B. Mulloy and S. J. Perkins, J. Biol. Chem., 2011, 286, 24842–24854 CrossRef CAS.
- N. Volpi, Chondroitin Sulfate: Structure, Role and Pharmacological Activity, Academic Press, Amsterdam, 2006 Search PubMed.
- R. Stern and M. J. Jedrzejas, Chem. Rev., 2008, 108, 5061–5085 CrossRef CAS.
- V. Gargiulo, M. A. Morando, A. Silipo, A. Nurisso, S. Perez, A. Imberty, F. J. Canada, M. Parrilli, J. Jimenez-Barbero and C. De Castro, Glycobiology, 2010, 20, 1208–1216 CrossRef CAS.
- J. W. Park and B. Chakrabarti, Biochim. Biophys. Acta, Gen. Subj., 1978, 541, 263–269 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |