 Open Access Article
Open Access ArticleDegradation of polymer films
David
Cheneler
*a and
James
Bowen
b
aSchool of Mechanical Engineering, The University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: d.cheneler@bham.ac.uk; Fax: +44 (0)121 414 7484; Tel: +44 (0)121 414 4256
bSchool of Chemical Engineering, The University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: j.bowen.1@bham.ac.uk; Fax: +44 (0)121 414 5377; Tel: +44 (0)121 414 5080
First published on 24th October 2012
Abstract
In this review paper the current state of research into the physical degradation of polymer films is elucidated. Modern applications of polymer films and the implication of their degradation are discussed. Recent investigations into solid interactions such as abrasion, adhesion, fatigue and other failure modes as well as plasma and photonic interactions are examined. This investigation highlights key degradation mechanisms as well as areas where controversy over these mechanisms lies, and suggests directions for future research.
 David Cheneler | David is a chartered mechanical engineer who received his MEng from the University of Birmingham in 2005 and his PhD in microengineering and microrheology from the University of Birmingham in 2010. His research interests lie in the development of sensor technologies for the electro-mechanical analysis of complex and biological tissues. |
 James Bowen | James received his PhD in nanoscale surface chemistry and adhesion from the University of Birmingham in 2006. His research interests lie in the areas of micro/nano-technologies and the application of multiscale metrological techniques to the characterisation of complex and novel materials, including their tribological, rheological and mechanical behaviour. He has a particular interest in atomic force microscopy, interferometry, and ellipsometry, as well as Raman and X-ray photoelectron spectroscopies. |
1. Introduction
The degradation of polymers and plastics is a vastly interesting and technologically important field. Thin films of polymers and plastics are worth special consideration, separate from studies of bulk materials because there are many phenomena that occur in thin films that are relatively unimportant in bulk polymers, but result in radically different structural properties.1,2 As films get thinner and approach length scales comparable to the radius of gyration of the polymer, the influence of surface effects becomes more significant.3,4 Besides specific surface modifications, this is due to the lack of neighbouring molecules at the surface as compared to the bulk.3,5 This results in a change of the number of possible chain conformations at the surface and an enrichment of chain ends due to entropic effects. The changes of mobility and chain kinetics are evident at both the free surface and at the substrate–polymer interface.5,6 Even below the melting temperature, polymers tend to form a very thin mobile, quasi-fluid surface layer close to the substrate surface due to the reduction of entanglement.6,7 This confinement and the subsequent organization and reduction in number of conformations of the polymer next to a solid interface also increases the number of possible cross-linking sites. As the elastic modulus increases with an increased cross-linking density, a thin film may have a larger modulus than the bulk material at the same density and molecular weight.8 In polymers cross-links may take two forms: chemical which are covalent in nature and physical. Physical cross-links can involve (i) polymer chain entanglement, (ii) bridging species between polymer chains (e.g. cations between anionic moieties on polymer chains, such as alginate) and (iii) hydrogen bonds between polymer chains, between moieties which are not charged, but where one moiety is electron-rich (e.g. O in H2O) and the other moiety is electron-deficient (e.g. H in H2O) due to electronegativity differences between local atoms.The exact definition of a thin film is a problem of semantics and is dependent on the field one is in and the purpose of the film. A film is a thin layer or membrane of any material. The film can be considered thin if its thickness is much smaller than all the other critical dimensions of both the film and phenomena that act upon it. This means a thin film will have a thickness small relative to its radius of curvature or its extent; it will be less than the diameter of abrasive particles, the wavelength of radiation that acts upon it, for instance. In the literature, polymer films are generally considered thin if they are of the order of a few microns thick or less.9 This is the definition generally assumed in this paper. However, in order to be relevant to the greatest number of industries, a few extreme examples have been described which are slightly outside this definition but are considered thin in the industry that uses them, in particular agricultural films and aeronautical coatings. Naturally not all degradation processes will occur with equal significance in films of different thicknesses hence this review has attempted to describe these processes in a general a manner as possible given current levels of understanding.
In films that are thin, surfaces have a decisive influence on the physical properties of the structure, whereas in thick films it is the bulk properties that dominate. Besides the inherent differences they have with bulk materials, polymer films can also be modified to have a patterned surface chemistry useful in biological and electronic research.10 Patterns can be generated using parallel techniques such as photolithography,11,12 micro- contact printing13,14 and imprint lithography15,16 and by ‘direct write’ techniques including electron beam lithography (EBL),17 dip pen nanolithography (DPN),18–20 laser ablation21 and near-field scanning optical microscopy (NSOM).22
Modification of the properties of polymer films is also achieved through the use of additives and the formation of composites which can have special significance given the comparable length scales of the polymer film and additive. Composites can be made in a variety of ways in order for them to have specific properties. The main methods include mixing the polymer melt directly with the nanoparticles (melt mixing), dissolution of the polymer and particles in an adequate solvent with evaporation of solvent (solution method) and mixing of particles and liquid monomer followed by polymerization (in situ polymerization).23 Additives are generally divided into two types: extenders, which are insoluble particles introduced into the polymer matrix in order to occupy space, and functional fillers such as stabilisers and sensitizers. Stabilisers include light absorbers such as carbon black,24 UV absorbers such as dihydroxybenzophenones and benzotriazoles,25 anti-oxidants including phenols,26 quenchers which absorb electronic excitations and include metal chelates,27 radical scavengers28 which donate hydrogen atoms, such as quinones and polynuclear hydrocarbons, peroxide decomposers which donate electrons,29 and include alkyl xanthates, and nucleating agents which reduce chain mobility and diffusivity of attacking sites – these include metal salts and bis-phenol phosphates. Sensitizers include metal oxides whose activity is related to their semiconductor zones,30 metal salts which form ions and carbonyl compounds and dyes like anthraquinone which generate singlet oxygen.30
The addition of fillers to bioresorbable polymers can be used to alter the polymer degradation behaviour, by allowing rapid exchange of protons in water.31,32 Polymer films can also be used as a scaffold allowing the nanoscale properties of other materials to be taken advantage of. In particular, nanoparticles of noble metals exhibit significantly distinct properties from their bulk counterparts.33,34 This permits polymer composites with unique properties such as plasmon absorption,35 near-IR photoluminescence36 and superparamagnetism.37
1.1 Modern technologies enabled by the development of thin polymer films
Thin polymer films have recently enabled a number of novel technologies to be developed, advancing several fields. For instance within electrical engineering, electrical components such as thin-film polymer transistors incorporating spin coated gate dielectrics and conducting sources have been improved significantly resulting in low cost, large area, even flexible, electronics.38 Applications such as active-matrix displays, ink jet printing and integrated circuits, in particular radio frequency identification (RF-ID) tags, are becoming more common.39 Typical materials include spin-coated poly-4-vinylphenol (PVP) as the organic dielectric gate material and polyethylenedioxythiophene (PEDOT) doped with polystyrene sulfonic acid (PSS) as the conducting polymer.40 While it is known that solid polymer electrolytes (salt dissolved in a polymer matrix) suffer from slow polarisation response, newly developed polymer-ion gels made using ultra-thin cross-linked polymer films, either from nanometre thick self-assembled monolayers or thicker high-dielectric ferroelectric films, have been shown to have specific capacitances that exceed that of conventional ceramic or polymeric gate insulators in organic thin-film transistors.41 The films in this study were formed by the gelation of a triblock copolymer, poly(styrene-block-ethylene oxide-block-styrene) (PS-PEO-PS) (7 wt%), in the ionic liquid 1-ethyl-3-methylimidazolium, but it was recognised that a convenient deposition method such as ink jet printing of high-capacitance dielectrics that serve as gate insulators is still a challenge.41Also, owing to the excellent electrical insulation properties of some polymer films, such as bi-axially orientated polypropylene (BOPP), polymer films are often used as the dielectric in capacitors and in other electronic components.42 They are also used as encapsulants and sealants.43 Also the next generation of solar cells are being fabricated using thin polymer films.44,45 Semiconducting polythiophene (PT), an insoluble and difficult-to-process polymer, has been deposited using oxidative chemical vapour deposition and applied as an electron donor in bilayer heterojunction solar cells.44 In an alternative design, poly(3-hexylthiophene) (P3HT) and [6,6]-phenyl-C-61-butyric acid methyl ester (PCBM) was deposited using a dip coating method to obtain the active layers of a solar cell.45
In medical science and biology, cells are grown on biodegradable polymer films and scaffolds to promote tissue growth and remodelling. The polymers degrade and are absorbed by the body; therefore it is critical that the polymers used are biocompatible with good mechanical strength and surface chemistry that allows for the proliferation, differentiation and adhesion or migration of cells.46,47 Polymers used in these applications can in general be divided into two camps: synthetic and natural polymers. Biodegradable synthetic polymer materials such as poly(glycolic acid) (PGA), poly(lactic acid) (PLA), and their copolymers, poly(p-dioxanone), and copolymers of trimethylene carbonate and glycolide have been used in a number of clinical applications. Natural biodegradable polymeric materials are derived from proteins such as collagen, gelatin, and albumin and polysaccharides such as cellulose, hyaluronate, chitin, and alginate.46,47 Comparable bioresorbable and biodegradable polymer–drug matrices are also being increasingly used as a coating for in vivo medical implants such as drug release systems, sutures and orthopaedic implants.48,49 They allow for the maintenance of drug levels without resorting to repeated administration of drugs as reabsorbable material can be broken down and the degradation by-products eliminated from the body. In particular polymers with ester linkages in the main chain are ideal for temporary biomedical devices.48–50 A technology recently utilised in the medical sciences to fabricate these polymer matrices and other coatings are polyelectrolyte multilayer (PEM) coatings.51–53 They are used to alter the surface energy of a material by controlling the concentration of polyanions and polycations.51 This has particular application in tissue engineering where it can be used to affect cell adhesion and mobility by controlling protein adsorption.51–53
Thin polymer films have also been utilised for drug delivery in coacervates and nanocapsules. Coacervates are spherical colloidal droplets of assorted organic molecules, such as proteins, with diameters typically in the range of 1–100 μm.54,55 They are formed by dispersing the core material in a solution of the coating polymer which is dissolved in a solvent. The three immiscible phases are physically mixed and the liquid polymer adsorbs at the interface of the core droplet before it is rigidized by cross-linking techniques. These microspheres or microcapsules are used mainly in the food and medical industries to affect taste and texture in food56 and also as a drug delivery mechanism as an alternative to foams comprised of hydrogels such as gelatin and alginate.57 Related to coacervates, but at a smaller scale, are polymer-based nanocapsules, which are often used for drug delivery.58 They are typically 100–500 nm in diameter but can range from 5–1000 nm. They can be defined as a nano-vesicular system that exhibits a typical core–shell structure in which the drug is confined to a reservoir surrounded by a polymer membrane or coating. They can be fabricated in a number of ways including that used by coacervates and by using PEMs. Examples of active agents include xanthone, DNA and insulin.59,60
In microengineering, especially in microfluidics, thin polymer films are used to protect structures from chemical attack and to facilitate fluid flow by increasing interfacial slip61 or assisting electrophoresis,62 and can also be used as a structural component such as membranes in valves and pumps.63 These microfluidic channels are commonly fabricated from poly(methyl methacrylate) (PMMA) and poly(dimethylsiloxane) (PDMS).61,64 It has been shown that by coating the polymer substrates with PEM coatings such as poly(diallyldimethyl ammonium chloride) and poly(styrene sulfonate) (PDADMAC/PSS) or chitosan/alginate renders the surfaces hydrophilic and decreases organic solvent penetration, preventing the swelling of PDMS due to chloroform exposure, for example.62,64
Other applications for thin films include biodegradable thin polymer films used in agriculture as a cover for crops to provide protected cultivation and a better micro-environment for plants as well as protecting against adverse weather conditions.65 This makes agriculture more sustainable and less reliant on herbicides and pesticides, and makes water conservation more efficient. They can be employed to create low surface energy surfaces, fluorinated polymers such as octafluorocyclobutane and Teflon are used in particular. These can be applied to surfaces to increase hydrophobicity and reduce friction for a wide variety of applications.66 Polymers films are also used to protect surfaces from chemical attack. For instance, PMMA and related copolymers have been used to coat stone and protect buildings and monuments from pollution and acid rain.67
1.2 The state of research into the degradation of thin polymer films
The degradation of thin polymer films is of significant importance as it can cause a deleterious change in the chemical structure and in the mechanical properties of the film. As will be made clear later there are a wide variety of different degradation mechanisms caused by interactions between the polymer film and its environment. There are many degradation processes caused by a wide range of environmental factors, including photodegradation by light, typically in the UV wavelength range, biodegradation through the action of micro/macroorganisms and related biological agents, high-energy degradation by exposure to radiation such as X-rays, γ-rays, and β-rays, chemical degradation through hydrolysis, solvolysis, decomposition and/or etching, mechanical degradation in the form of abrasion, adhesion and fatigue failure, thermal degradation usually due to exposure to high temperatures, oxidative degradation, and corrosive degradation due to plasmas.The degradation of thin polymer films is a topic worthy of study, not just out of academic curiosity, but because of the wide range of ‘real’ scenarios in which degradation can have a significant influence, whether it is advantageous or disadvantageous. Degradation is advantageous when the polymer needs to be disposed of, for instance. A serious side effect of polymer films being increasingly used in agriculture concerns the growing disposal problem of thousands of tons of agricultural plastic waste, which are frequently burnt in uncontrolled fires.65 The advantages of these films will only outweigh these issues if the films can achieve good mechanical behaviour throughout their useful lifetime and be 100% biodegradable in the soil after the end of its lifetime, preferably before the next cultivation season.65 Biodegradable polymers in medical devices can be used to prevent conditions such as stent thrombosis by careful release of the appropriate drugs.49 In these cases, the rate of biodegradation must be sufficient to release effective doses of the drug, but the polymer has to be mechanically stable during the whole period of treatment. The ability of a polymer to degrade when needed also has application in microengineering as polymer films can be used as a sacrificial layer during fabrication of micro electromechanical systems (MEMS).68
Degradation is not always desirable. It is well known that degradation of polymers can lead to reduced performance and the limited lifespan of devices in which these films are employed. For example, degradation or hydrolysis of polymeric components, especially in semiconductor applications, is a source of undesirable species that can cause electromigration and can reduce device reliability and lead to device malfunctioning.43 Degradation can also have more severe implications. Polyester, for instance, is an important polymer used in medical implants and coatings and is comprised of monomers linked by ester moieties. However, esters of fatty acids occur naturally and it has been observed that degradation of these medical implants via routes involving the breakdown of ester bonds has previously unconsidered medical effects such as disrupted pancreatic beta-cell function and increased insulin resistance.69,70
Compared to other research areas such as metals, coatings and even plastics, there is inadequate understanding about the degradation of thin polymer films, with research limited in general to very specific cases. The extent of this issue is reflected in the literature. A cursory search on Web of Knowledge in March 2012 (ref. 71) shows that at that time, a total of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 136011 papers have been published on either degradation or wear in journals in that database. This covers over 130 years of research from fields as diverse as engineering, agriculture and microbiology. The vast majority of these papers however, are very much concerned with chemical stability at a molecular level and not with the degradation of solids. Out of the papers that are concerned with solids, 40491 concentrate on metals with a great deal of important studies occurring in the fifties and early sixties. 13199 papers concentrate on ceramics with particular emphasis on the effects of coatings for cutting tools for machining and drilling tools for the petroleum industry and wear in prostheses and bone replacements. The major fundamental studies originated in the sixties and seventies. As well as these, there are over 48175 papers on polymers. However, the majority of these papers are on the kinetics and degradation of liquid polymer melts with only 11696 actually on solid polymers and plastics. Three quarters of these papers were published in the 21st century although there are some very important studies which focussed on weathering and thermal degradation in the fifties. Naturally most of these papers concentrate on bulk plastics with only 1558 papers published on the degradation and/or wear of thin polymer films. While the first papers appeared in the seventies, 1183 were published within the last ten years. It is clear what the impetus was for the study of degradation in thin polymer films is. While a number of fundamental studies were carried out for their own sake,72,73 the two main drivers were the medical74,75 and microengineering/computing industries.76,77 In both fields, the studies were based exclusively on the applications and devices based on thin polymer films with a view to optimise specific designs rather than to develop a better understanding of degradation.74–81
136011 papers have been published on either degradation or wear in journals in that database. This covers over 130 years of research from fields as diverse as engineering, agriculture and microbiology. The vast majority of these papers however, are very much concerned with chemical stability at a molecular level and not with the degradation of solids. Out of the papers that are concerned with solids, 40491 concentrate on metals with a great deal of important studies occurring in the fifties and early sixties. 13199 papers concentrate on ceramics with particular emphasis on the effects of coatings for cutting tools for machining and drilling tools for the petroleum industry and wear in prostheses and bone replacements. The major fundamental studies originated in the sixties and seventies. As well as these, there are over 48175 papers on polymers. However, the majority of these papers are on the kinetics and degradation of liquid polymer melts with only 11696 actually on solid polymers and plastics. Three quarters of these papers were published in the 21st century although there are some very important studies which focussed on weathering and thermal degradation in the fifties. Naturally most of these papers concentrate on bulk plastics with only 1558 papers published on the degradation and/or wear of thin polymer films. While the first papers appeared in the seventies, 1183 were published within the last ten years. It is clear what the impetus was for the study of degradation in thin polymer films is. While a number of fundamental studies were carried out for their own sake,72,73 the two main drivers were the medical74,75 and microengineering/computing industries.76,77 In both fields, the studies were based exclusively on the applications and devices based on thin polymer films with a view to optimise specific designs rather than to develop a better understanding of degradation.74–81
This trend of focussing on specific applications rather than fundamental science has continued to this day and has led to a situation where degradation in thin polymer films is incompletely understood with many theories on degradation mechanisms remaining controversial.82–86 There are a number of reasons why degradation in polymer films is incompletely understood. The main reason is simply the sheer number of applications and compositions of thin polymer films. The number of polymers available, the number of substrates, types of fillers and additives, deposition methods and the range of thicknesses that are possible results in a bewilderingly large array of possible combinations to consider. For engineers and scientists, on one hand this is perceived to be an excellent situation as it seems that a thin polymer film can be designed to fit any purpose. But with such a wide range of different compositions, chemistries and structures available within a thin polymer film, it is difficult to form categories of films in which particular failure modes and/or degradation mechanisms will occur in a specific situation. Without this, the understanding of degradation in thin polymer films will remain fragmented and insufficient to be used to predict the performance of new films. The expansiveness of the field is not the only reason. In essence, the need to understand degradation in modern polymer films is a new need due to recent developments in the field. A lot of new polymer films are still firmly within the realms of the prototype used for research purposes and hence there has generally been little consideration of the products life cycle and its long term performance. However, with technologies maturing and being commercialised, this is no longer feasible. The length scales inherent in the study of thin polymer films is also an issue. The resolution of measuring technologies, such as tribological equipment through to spectrometric analysers, has had to be improved significantly in recent years in order to give any form of meaningful data on thin polymer films. Without this data, early studies have been only able to hypothesise about the degradation of polymer films. The lack of data has also prevented the complete mathematical analysis of the different degradation mechanisms. This is particularly apparent when considering solid interactions. Given that contact mechanics has been studied for over a century, it seems odd that friction, wear and plastic deformation in polymer films is still poorly understood with many competing theories.84,87,88
1.3 Content of paper
The purpose of this paper is to review the state-of-the-art research into the degradation of thin polymer films. Degradation processes due to solid, plasma and photonic interactions will be described with a view of elucidating what the current opinions are in these areas and suggest where further research is required. There is naturally a large overlap between the mechanisms that cause degradation in bulk polymers and those in thin polymer films. Research into the degradation of bulk polymers has a longer history and is more comprehensive, and many quality reviews have already been written in this area.23,84,86,87,89–91 This paper focuses on films and effort has been taken to concentrate on pertinent degradation mechanisms found specifically in these systems rather than discussing all possible mechanisms in a generic fashion. It will be noted that liquid interactions are not included. This may be an odd omission given the large array of industrially pertinent situations, such as the effect of local pH in sea water on the degradation of polymer coatings on sea vessels,92,93 for instance. This omission is deliberate due to the near infinite number of polymer–chemical combinations that are possible, see references for examples.91,94–99 It is certainly too vast a subject to be treated comprehensively in a mere section of a review paper. As these interactions occur on the atomic length scale, they are in general independent of the thickness of the polymer and so are not specific to the systems of interest here, even though chemical interactions are frequently used to modify and pattern thin films.100,101 Exceptions may include instances where the polymer film is insufficient to protect the substrate resulting in etching and loss of adhesion.102,1032. Solid interactions
2.1 Wear mechanisms
The most common wear modes of polymers due to solid interactions are abrasion, adhesion and fatigue.88,104–108 However, given the interrelation between the different mechanisms, rigorous classification of wear processes has been recognised as being impossible, even within a narrow range of contact variables such as slider geometry, applied load, slider velocity and temperature.87,104–108 As a coarse definition, abrasion occurs when a harder material acts on the polymer film causing scratches or gouging. This can be due to plastic grooving (see Fig. 1a), or ploughing (see Fig. 1b), in which material is pushed ahead of the particle and displaced sideways to form ridges adjacent to the groove.106 In this case, no material is removed from the film. When the abrasive particle cuts the film, all material displaced is removed as a chip (see Fig. 1c). Sometimes adhesive junctions between the particle and polymer form due to localised bonding.87 When the particle is moved relative to the polymer, this junction causes a transfer of material from one surface to another in a manner depending on polymer properties and local conditions especially frictional properties and flexibility of the chain molecules of the polymer. Fatigue is due to repeated or cyclic stressing resulting in an accumulation of irreversible changes in the form of crack formation and progressive fracture.108 These wear mechanisms are particularly important due to the use of thin polymer films to reduce the frictional properties of components such as bearings.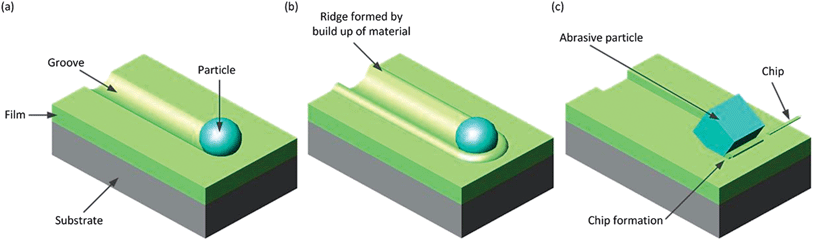 | ||
| Fig. 1 Some wear mechanisms due to solid interactions. (a) Plastic grooving in which material is plastically compressed beneath the slider, (b) ploughing when material is instead pushed aside to form ridges and (c) abrasion where all displaced material is removed as a chip. | ||
For instance, PTFE has a low coefficient of friction which makes it a useful bearing material but suffers from a high rate of wear.109 When wear occurs during interaction with a hard surface, the PTFE chain undergoes scission, creating active bonds which chemically react with the countersurface. This results in strong adhesion, material transfer, and formation of a more defect-free film on the countersurface. Subsequent interaction between the original film and the transfer film causes anisotropic deformation of the unit cell, which results in closeness of adjacent chains and easy shear between chains. This rearrangement and alignment of the polymer chains causes growth and reorientation of existing crystallites situated in a very thin subsurface region of the polymer film.109
It is important to recall that it is not just the chemical structure of a polymer chain that affects its properties but also the structure, i.e. orientation, of the chain in situ. One of the primary factors affecting the resultant structure is the method of deposition. For instance Iwamori et al. showed in a study where PTFE was deposited using three different types of R.F. sputtering (in this case via a magnetron) and tested using the pin on disc technique, that both wear resistance and coefficient of friction increased with the magnetic field strength of the sputterer.110
Tests conducted using a friction abrasion analyser showed that the hydrophobicity of fluorinated carbon film coated surfaces degraded rapidly with wear. As these films have a low surface energy, they generally have low coefficients of friction which are usually desirable. However, this low surface energy also results in poor adhesion between the polymer film and substrate causing rapid film removal with wear. Beckford et al. showed that it can be necessary to generate a large surface roughness on the substrate prior to deposition of the polymer to maintain hydrophobicity during wear processes.66 This process does not improve adhesion of the film, but rather uses the higher hardness of the substrate to protect the polymer film from wear.
Organic thin films such as ultra-high molecular weight poly(ethylene) (UHMWPE), which has a linear structure, are also used to reduce the coefficient of friction, adhesion forces and wear. Satyanarayana et al. prepared 1.4 μm thick films on silicon substrates using dip coating.111 However it was observed that adhesion was poor between the substrate and polymer film. They used chemisorption with an intermediate self-assembled monolayer (SAM) of 3-glycidoxypropyltrimethoxysilane which resulted in higher adhesion and better wear durability. Specifically it was shown that the films with intermediate layer had a wear life of 100000 cycles at load of 300 mN compared to 12000 cycle life at 70 mN for the single layer films.111
Wear particle-induced aseptic loosening of hip replacement prostheses remains a major cause of revision surgeries for the commonly used metal–UHMWPE total joint arthroplasty.112 Surface modification, through the application of coatings, offers the potential to reduce the wear rate without compromising the bulk mechanical behaviour of the implant material. One method used to significantly increase the wear resistance of orthopaedic implants is to coat the substrates with PEMs (polyelectrolyte multilayers). The tribological properties of PEMs comprised of poly(acrylic acid) (PAA) and poly(allylamine hydrochloride) (PAH) were tested in both ambient conditions and in the presence of a liquid medium in the form of bovine calf serum.113 These tests consisted of 2000 cycles of reciprocating motion under normal stresses of up to 450 MPa applied using flexure-based biaxial testing apparatus with a flat-on-flat configuration. During the tests, there was significant wear prevention for steel, glass and ceramic substrates in the dry state compared to uncoated substrates. It was observed that delaminated film fragments were responsible in preventing contact between the mating surfaces. In the presence of the bovine calf serum, substrate wear was avoided over 30 cycles with only 70 nm-thick PEM films.113 In a follow-up study, PEM coatings, a few hundred nanometers thick, on both metal–metal and metal–UHMWPE systems were exposed to over 500000 cycles of bi-directional motion in a macroscale pin-on-disk test.84 In the latter case, the use of the films reduced UHMWPE wear by up to 33% when compared with the uncoated control. This is the first clinically relevant laboratory demonstration of the wear-reducing ability of these films.
2.2 Other failure modes due to solid interactions
Delamination of polymer thin films occurs when the induced stress due to indentation/sliding becomes comparable to the critical buckling stress.114,115 This happens when the contact area between two bodies is small, leading to high strains. In the ideal case, this occurs when the elastic energy is comparable to the true (thermodynamic) work of adhesion required to create two new surfaces at the expense of the interface. In real situations however, de-adhesion that results in delamination is also affected by inelastic damage, plasticity, viscoelasticity and micro-cracking occurring in the regions of the substrate and film near the interface (Fig. 2). Essentially any dissipative mechanism will affect the energy involved in the process. It has been observed that buckling phenomena are particularly significant in thin film systems where the substrate has a lower modulus than the film. Incidentally Kriese et al. reported that comparable data for the interfacial toughness of phenol-formaldehyde, a brittle polymer, was found using indentation as well as scratch tests.114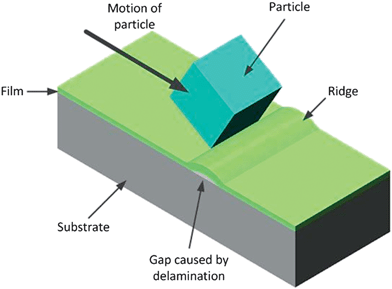 | ||
| Fig. 2 Possible delamination process. Note the similarity to Fig. 1c, the most likely difference causing the different outcomes being relative adhesive to cohesive strengths of the materials involved. | ||
Delamination can also occur due to thermal stresses. The heat energy can come from a number of sources including dissipation of phonons due to induced vibrations caused by stick-slip phenomenon during sliding,116 due to particle impingement115 or exposure to electromagnetic radiation, especially in the infrared spectrum.117 These thermal stresses can cause a thermal gradient in a two layer composite system that leads to bending and permanent plastic deformation after cooling. Thermal expansion mismatch can also result in buckling delamination, not only due to a mismatch with the substrate, but also due to a mismatch with an oxide or glassy, crystalline top surface.116 Buckling is a mechanical instability whereby a flat film develops out-of-plane undulation. This happens when elastic thin films are attached to a substrate which imparts a compression, perhaps due to thermal mismatch, and above a critical load can cause wrinkling.118 The effect of residual stresses and surface deformations become significant as the thickness of the film approaches nanoscale. Other failure modes that are possible include wrinkling, tunnel cracking and interface debonding.119,120
Besides stress-induced adhesive failure of the film to the substrate, cohesive failure of the film can also occur. Polymers are generally bonded to the substrate through van der Waals interactions and hydrogen bonds.121,122 The hydrogen bond forms with polymers when the hydrogen atom is linked with an electronegative atom resulting in a common proton forming a strong and stable bond under favourable conditions. Electrostatic attraction makes a contribution to the adhesion of polymers when an electric double layer is formed owing to a transfer of electrons from one surface to another. The direction of the transfer depends on the substrate. If the substrate is metallic, for instance, it will be an electron donor. If the interfacial bonding is stronger than the cohesive strength of the polymer layer, than the polymer will be fractured and polymer transfer will take place. As a rule, in polymers, the interfacial forces and forces acting between polymer chains are nearly equal, cross-linking bonds are usually hydrogen bonds in nature, and fracture often occurs in the polymer itself.87
2.3 Effect of fillers
Mechanisms of wear and failure become even more complicated when composites are considered. The combination of inorganic–organic fillers and polymer matrices at the nanometre scale (nanocomposites) has become an efficient strategy to improve the structural and functional properties of the original polymer.31,123 One of the most attractive features of nanocomposites is their ability to more effectively emulate organic tissue.31 For instance, natural bone is an organic–inorganic composite material comprised of collagen and apatites. However, introducing fillers and additives also introduces new potential failure modes in the polymer film, the major factor being the interfacial adhesion between the nanoparticles and polymer matrix. This adhesion, as well as the properties and distribution of the fillers, have a significant effect on the mechanical properties of the composite.123Nanocomposites are used for improving triboengineering components such as gears, bearings and other parts covered in thin polymer films.124,125 In this guise the composites form solid lubricants and contain fillers such as layered silicates, or clays such as montmorillonite, hectorite and saponite, graphite or molybdenum disulphide.126 Many polymers have been investigated as potential matrices for nanocomposites, the most common being polyamides and polyesters such as PET, poly(ethylene terephthalate) and PBT, poly(butylene terephthalate). These composites promote the formation of transfer layers on the countersurface and decrease the coefficient of friction. These films typically fail because of fatigue. Fatigue causes pitting, internal delamination between filler and matrix, and formation of wear debris as a result of propagation and intersection of small surface cracks orientated perpendicular to the sliding direction. In thin polymer films the frictional properties may be velocity dependent owing to local increases in temperature. It has been observed that nanoparticles fillers can significantly increase the thermal resistance and the heat deflection temperature (HDT), the temperature at which a polymer deforms under a specific load, of polymer films. Kojima et al., for instance, noted that incorporation of the silicate Montmorillonite increased the HDT of Nylon 6 from 65 °C for the pure polymer to 152 °C.125 It has also been noted that the increase in thermal resistance not only makes the frictional properties more consistent, providing failure doesn't occur, but also increases the resistance to thermooxidative degradation.122
Microcomposites generally result in deleterious wear properties as compared to pure polymers and nanocomposites. As micron sized particles are significantly larger than the surrounding polymer chain segments, they tend to get gouged out easily, resulting in significant material removal during sliding.127 The particles used in nanocomposites, however, are of a similar size to these segments and so can help reduce wear. The tribological behaviour of these materials is complex. A number of tuneable parameters need to be optimised to control friction and wear damage. Important parameters include size, aspect ratio, concentration and orientation of the particles. The relative hardness and interfacial adhesion are also important considerations.
Lee et al. investigated the tribological effects of carbon-based nanoparticles in PTFE films.128 Using a pin and disc type wear test, it was observed that the addition of 2 wt% of carbon nanoparticles reduced the wear coefficient of PTFE to a quarter of its value for the pure polymer. Several similar studies have been performed on bulk nanocomposites.127–129 While most show enhanced wear resistance as compared to pure polymers and microcomposites, results vary widely from study to study. Also, due to the number of different property combinations and the range of testing parameters possible, there is no real consensus on the mechanisms of wear resistances in nanocomposites.
It has been noted that in general there is poor characterisation of polymer nanocomposites and a lack of comprehensive quantitative descriptions of observed phenomena in the literature.84 It was also observed that the range and deviations in wear rates and scratch penetration depths are not sufficient to compare materials. This shortcoming has led to false impressions of observed phenomena.84
2.4 Analysis of solid interactions
Typically, the elastic moduli and hardness of thin films such as these are tested using indentation, for which both nanoindenters and AFM are used.130,131 In most cases Hertzian contact mechanics theory is employed, with variants used depending on the type of probe used, i.e. spherical probes or Berkovich indenters which exhibit a three-sided pyramidal geometry, for instance.132 Excellent reviews on this technique exist.132,133 However this technique is limited for very thin films because if the stress field due to indentation is of the same depth as the film thickness, the modulus of the substrate influences the measured data.134 Essentially the substrate will impart another stress field onto the field due to indentation as a result of the reaction force to the compression. If the film is sufficiently thick or the indentation depth is sufficiently small these fields will only weakly interact and the Hertzian theory will be reasonably accurate. Otherwise the presence of the substrate needs to be taken into account. Classical theories of indentation are based essentially on Sneddon's analysis, but in these derivations it is assumed that the material is an infinitely large incompressible specimen.135,136 Some excellent mathematical analysis has taken place, especially in the 1960's and 1970's to extend the analysis to the thin film case. Analyses tended to concentrate on axisymmetric problems such as punch or spherical indentation, generally assuming that the punch and substrate were rigid with an elastic layer in between,134,137 although multi-layered elastic systems have been considered.138–142Many techniques have been developed for the elastic and elastoplastic indentation of thin films.142–145 Elastoplastic materials have been extensively studied as the remaining permanent deformation of the film allows for post-processing, thus yielding more information. It has been shown that naïve indentation tests may be insufficient as many dissimilar materials may still yield similar indentation curves and produce non-unique solutions.146–149 However, generally these techniques are only applicable to crystalline materials and so are not necessarily appropriate for most polymers. In general, models will only apply to ideal films. In more complex cases where failures may occur in the form of the debonding150 or cracking of the polymer film, or when non-isothermal cases are considered,151 more analysis is required.152
Indentation of viscoelastic materials, which polymers often are, has proven to be more technologically challenging given the sensitivity and quantity of transient data required. It is also a more mathematically challenging problem because of the difficulty of determining the correct constitutive equations that properly describe the polymeric nature of the material. Due to the equivalent problem in analysing biological materials numerous indentation techniques have been developed, focussing primarily on oscillatory indentation153,154 or creep155/stress relaxation experiments.156,157 The viscoelasticity of the films leads to a complex transient stress state that depends not only on the current loading but on the memory of past loading states.153,155–158 It has been shown that viscoelastic solutions can be gleaned from the elastic solutions by way of building in memory effects through the correspondence principle.158–161 However, most of the mathematical expressions of the stress states during indentation described above are not in a convenient form to apply this principle. Fortunately, the series solution derived by Dimitriadis et al.134 are in a convenient form and have been used to derive viscoelastic solutions for limited cases.162
The stress distribution is even less understood in sliding contact. This is important for the study of scratch tests and is an area of intense study. Fundamental knowledge about mechanical behaviour during scratching is not nearly as developed as indentation testing owing to the complexity of the boundary value problem. Not only is the problem mathematically complex but complications also lie not only in the need to use more general constitutive equations that properly reflect failure modes such as elastic-plastic scratching and delamination. Fundamental studies by Archard in the 1950's and 1960's generated frictional relationships for sliding wear in metal systems but many of these relationships cannot be applied to polymers due to their generally non-isotropic and viscoelastic nature.163–166 Classical theories suggest that the friction force in an unlubricated contact does not depend on sliding velocity.109,124 This is true if the contact temperature grows negligibly. As polymer films are viscoelastic to some degree, a number of relaxation processes are possible and heat dissipation is complicated being a function of the physiochemical activity of the polymer. As a result, contact temperature may be significant leading to a complex dependence of the friction coefficient on velocity.88,109,124 This dependence is particularly significant when in the course of testing, the temperature approaches that of the polymer glass transition temperature, under which conditions segment mobility will increase. Below this temperature the mobility is frozen and velocity dependence is minimal.
Besides calculating stress fields, even determining penetration depth is a non-trivial issue as localised vertical displacement of material surrounding the scratch leads to inaccuracies in scratch depth estimation. Based on the Hamilton and Goodman model,167 higher tensile stresses behind the slider on the surface of higher modulus polymers during scratching should promote surface cracking and/or debonding between phases of the multiphase polymers. It has been shown that molecular structure and orientation of the polymer and the mechanical and thermal properties of the substrate play a significant role in affecting the wear behaviour.168–170 Numerous studies have been undertaken which concentrate mostly on numerical models and experimentation, but still there is no real consensus on what the most appropriate models to use for predicting behaviour of polymer thin films in general.168–182 One significant reason for this is that analytical equipment such as atomic force microscope, Raman spectroscopy, interferometry and scanning electron microscopy, etc. with the resolution to measure forces and image bond energies and topologies at this scale have only recently been developed.175 Typically however, while the individual techniques can yield good results, they can only give a partial view of the situation. Perhaps wear will only be understood when multiple data types can be measured from the same experiments simultaneously using composite equipment configured for this purpose.
3. Plasma interactions
It is well known that polymer surfaces are modified upon exposure to activated gas plasma and the process is frequently used to improve bondability and adhesion of surfaces.183 In particular, the surface energy is modified through the generation of ‘dangly bonds’ caused by unsatisfied valence electrons on immobile atoms which form free radicals on the surface of the polymer film. Immobilised free radicals are very reactive but more kinetically stable than mobile radicals due to steric hindrance and hence exhibit a longer lifetime. As plasma only affects the top-few molecular layers, the effects of plasma exposure are more significant for thin films.83The reactivity of the free radicals can result in both cross-linking and degradation of polymers.184 Chemical surface modification results when the species generated in the gas react at a surface to form stable products with physical and/or chemical properties that are different from those of the bulk. In many instances, etching and modification occur simultaneously.
By far, the gas most often used for plasma modification of polymers is oxygen, but other commonly used gases include ammonia, nitrogen, and fluorine-based molecules.185 Plasmas can have many different effects depending on the chemistry and energies involved, including removal of material (see Fig. 3), cross-linking and chemical reaction. Oxygen-plasma treatment can increase the surface energy of polymers, whereas fluorine-plasma treatment can decrease the surface energy and improve the chemical inertness. In particular C4F8 and SF6 plasmas are routinely used in microengineering to etch silicon as part of the Bosch process.185 Ion bombardment of a silicon based surface with fluorine plasmas is mostly a physical process with few chemical reactions possible. Ion bombardment in these cases only provide the activation energy necessary for etching reactions on the substrate surface where weakly bonded molecules are formed and subsequently desorbed into the gas phase, essentially becoming a sputtering process.186 In general an increase in surface energy is not observed with SF6 suggesting that it only causes structural modifications of the surface.
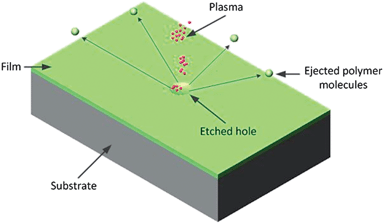 | ||
| Fig. 3 Depiction of the effect of directed plasma of a polymer surface. | ||
A large number of chemical reactions are possible in oxygen plasmas, and oxygen atoms are generally accepted as being the primary reactive species in initiating a modification reaction sequence. When polymer etching is performed at or near room temperature, polymer etching rates using oxygen plasmas in the absence of ion bombardment are much smaller than in the presence of ion bombardment. Etching in the absence of ion bombardment is often achieved with samples positioned downstream from the plasma, in a region known as the afterglow. Although there are no reports of differences in the rate of modification (e.g., based on wetting behaviour or surface composition) in these two conditions, structural differences such as degree of cross-linking of the modified polymer surfaces may result.187
Oxidation is generally unimportant for polymers that contain polar groups.184 For instance, it has been observed that there is no significant difference in the bond strength of cellulose acetate butyrate exposed with oxygen plasma compared to cellulose acetate butyrate exposed with nonoxidising plasmas, such as those formed from helium. However, poly(4-methyl-1-pentene) is another hydrocarbon which has two tertiary carbon atom groups per monomer unit which are relatively reactive therefore oxidation is important in this case resulting in significantly stronger bonds after plasma treatment.184
Alternatively, conductive films, such as poly(pyrrole), are porous and allow ion exchange with the surrounding medium. When the medium is an oxidative plasma, a multistep degradation processes can occur. In the case of poly(pyrrole) it was observed that chloride gas bubbles formed in the polymer which joined to form doughnut structures in the film and in turn circular features resulting in the thinning of the film.188
Modification of the surface energy is also used to improve the wettability of thin films and to change their hydrophobicity. Plasmas are frequently used for this purpose but the exact reason for the change in wettability depends on the polymer. Polyethylene readily cross-links under irradiation due to the formation of unsaturated groups which also results in improved wettability.184 Poly(styrene) does not readily cross-link under irradiation but still exhibits improved wettability. In this case this is due to the cleaning of the surface through the removal of organic matter.184
Besides material removal and surface modification, the use of plasmas in thin film modification can have deleterious effects due to higher concentration of reactive species as compared to exposure to gaseous medium. One example of this is in the fabrication of organic thin film transistors (OTFT).189 Common methods of patterning OTFTs include deposition through shadow masks, patterned growth or by using photoresist as an etch mask prior to etching with oxygen plasma.190 There are numerous advantages to the use of oxygen plasma, it is compatible with printing, photolithographic processes and allows for further encapsulation, but it has been shown to degrade transistor performance.
4. Photon interactions
4.1 UV exposure
Related to plasma exposure is exposure to electromagnetic radiation, especially ultraviolet light (UV). An example of a situation where this is important is the agricultural films used to cover crops in low tunnels discussed above. These are usually comprised of 30 μm thick low density polyethylene (LDPE) or poly vinyl chloride (PVC). Naturally these films are exposed to UV light via sunlight. However, a high dose of UV radiation was shown to have detrimental effects on the mechanical strength on these polymers.65 In particular the elongation of the polymer at break when stretch is applied, particularly when applied perpendicular to the polymer orientation, was significantly reduced. This was not necessarily the case in the parallel direction suggesting it is the cross-linking bonds that are most vulnerable to degradation due to UV exposure.Degradation due to UV exposure is generally due to C–C scission,191,192 resulting not only in chain scission but also photolysis and/or dissociation of side groups. This is a particularly important process with certain polymers, such as PMMA, cellulose acetate, etc. Many of the physical and chemical effects UV has on polymers can be explained in terms of either cross-linking or degradation. UV exposure as a degradation process is complicated by the fact that UV exposure can also be used to cure photo cross-linkable polymers, such as polyanhydrides, and improve mechanical strength.46 In these cases material degradation and mechanical properties are based on the choice of the monomer. The curing of macro monomers such as dimethacrylated anhydrides by ultraviolet light involves degradation by means of hydrolysis of the anhydride bonds.
Other complications arise due to the potential reversibility of the degradation. It has been shown using Raman and absorption spectroscopy that degradation of PMMA due to UV exposure can reduce over time.193 In this study it was shown that while the optical transmission of light through PMMA optical fibres was reduced shortly after UV exposure, its transmittance recovered to a significant fraction of its pre-exposure value after ten days. The change in optical absorbance of PMMA after UV exposure may be due to electron–hole trapping during the formation of photo-degradation products such as methane, hydrogen, carbon monoxide and carbon dioxide.191,192,194 They suggested a widely believed explanation for the UV-induced increase of attenuation that involves formation of different macro radicals which recombine progressively after UV is stopped.193
Aging in polymers exposed to ambient conditions occurs via oxidation due to free radicals in the polymer structure. This is particularly apparent in polymers deposited using PECVD. It was shown by Easton and Jacob that the uptake of oxygen is typically extremely rapid during the first few days after deposition associated with a high radical density, followed by a decline and eventual termination.195 In some cases, it was shown that the aging period can exceed one year for plasma generated polymers.196 This period is very dependent on the deposition parameters. It was shown through ellipsometric studies of thermal degradation that the thermal stability of plasma polymerised poly(L-lactide) (polyLA) can be increased by increasing the RF power.196,197
As mentioned previously, the durability of films has significant effect on the general economics or agriculture. There are also ecological implications as degraded plastics are frequently burned uncontrollably in the fields or dumped.65 It is necessary for polymer films to maintain its mechanical properties over its intended lifespan and then degrade away completely and harmlessly. Standard tests for UV exposure, or artificial aging tests, state that agricultural polymers should remain durable for 400 to over 7300 h of irradiance by UV at a wavelength of 340 nm at 0.35 W m−2. A study on 0.2 mm thick films of unstabilised LDPE showed a 60% reduction in tensile strength and increased brittleness and loss of strain-hardening behaviour.198 However, it was noted that standard tests do not accurately predict the life expectancy in the field and that weathering tests, under which real conditions impart the characteristics and properties of age, on the polymers need to be developed to ensure dependable results.
The degradation rates due to aging can be amplified if aging occurs parallel to repeated processing of the polymer, such as during recycling or remoulding of thermoplastics. It has been shown by Jansson et al. that combined degradation processes degrade faster than either extended aging or repeated processing performed separately.199 In this test, polypropylene (PP) powder was repeatedly compression moulded into thin films. The thermo-oxidative aging of PP results in hydroperoxide formation and decomposition into radicals resulting in polymer auto-oxidation. It was hypothesised that hydroperoxides formed during the ageing step, and decomposed in the subsequent processing step, thereby causing faster degradation of the polymer film. It was observed that the aging process substantially decreases the measured elongation-at-break for the polymers, but this could be restored with re-processing through re-crystallisation.
As yellowing and loss of colour is undesirable in the decorative coatings on aeroplanes, Varley et al. conducted accelerated weathering tests using UV-A fluorescent lamps on 1 μm thick high-gloss polyurethane monocoats.86 FTIR results indicated cleavage of amide groups and the growth of carbonyl groups from chain scission. This partial depolymerisation/decomposition due to low photon fluxes e.g. sunlight has been termed photo-yellowing.86 As UV is rapidly attenuated it was shown that an extra transparent coating with increased hardness and resistance to photo-activated cross-linking reactions significantly reduced UV degradation of the underlying film.82,86,200 This study observed that traditional weathering tests are not necessarily predictive of in-service performance. As actual stresses may include solar radiation, high temperature ranges (typically −50 to 80 °C), high localised temperatures due to hot air exhaust (∼ca. 120 °C), erosion from particulate matter, humidity and reactions with atmospheric pollutants such as sulphuric acid aerosol from volcanic eruptions, tests need to include combination of processes.
4.2 Laser ablation
Photon–polymer interactions can result in a variety of chemical reactions which range from modification of the polymer surface to complete degradation of the polymer. Ablation of polymers, or the removal of material due to a phase transition from its solid to its gaseous phase, by photon exposure usually occurs at high fluences such as that caused by lasers. Laser ablation can be used to pattern thin films by removing material201,202 or as a deposition mechanism.203–205 Deposition occurs when bulk polymers are ablated and the ejected gaseous monomers form a film on a new substrate.28,206 This kind of photon-induced reaction is limited to polymers that can depolymerise upon irradiation and decompose into fragments. A lot of common organic polymers, such as PMMA and PTFE, can be degraded by laser ablation which results in pyrolitic decomposition and/or carbonization of irradiated area depending on dose rate and duration.207 Particular polymers that can be depolymerised in this way are those formed by radical polymerization from monomers that contain double bonds.193 PMMA can depolymerise reversibly forming the monomer, hence its suitability.208 Polymers that are formed in irreversible chemical reactions that require the elimination of constitutive parts from their monomers cannot be formed into films through laser deposition. Polycarbonates and polyimides are examples of polymers that cannot be deposited in this way as they require the elimination of HCl or H2O respectively from their monomers during synthesis.82However, the choice of polymers is complicated by the fact there is no general agreement as to the exact mechanism of ablation.82,207 The two contending processes are photochemical and photothermal processes. Photochemical processes include direct bond rupture under electron excitation such as when organic molecules are excited into high-energy electronic states that decay through bond dissociation. Photothermal processes degrade cross-linking bonds and can cause chain scission through rapid conversion of photon energy into vibrational energy to produce heat. A number of studies have hypothesised that photothermal processes are dominant,209,210 but these studies were confined to a limited range of polymers. Other studies concentrating on organic polymer photochemistry have proven UV photons can cause photochemical reactions after irradiation of organic molecules; it is likely that photochemical reaction occur under ablation conditions.211–216
Almost all the photodegradation studies on PMMA with wavelengths in the shorter region of UV around 250 nm have led to a total or partial cleavage of the ester side chain of the PMMA molecule depending on the wavelength and energy of the laser whereas wavelength in the longer side of 250 nm have led to chemical reactions such photopolymerization, cross-linking or curing.217,218
It has been observed that the effect of radiation on a polymer depends on the radiation wavelength: the target is sputtered (ablated) under the action of the short-wavelength (300–400 nm or less) UV component, while the longer wavelength component causes heating and thermal damage of the interior of the film. In a study on the ablation due to an electrical arc in a low voltage circuit breaker comprised of polyamide-6 and PMMA,219 polymer vapour was observed. It was hypothesised that under most irradiation conditions, both mechanisms work simultaneously: primary bond rupture and formation of radicals are photochemical processes and further depolymerization and bond rupture are photothermal effects. The longer the action and wavelength, the more dominant the photothermal mechanism. It was also observed that polymer sputtering has a threshold in the energy of action and therefore begins in an explosive manner.219 For a near-threshold action, the removed mass linearly depends on the energy density. Intense sputtering is governed by absorption in the vapour (plasma), and the removed mass versus energy density dependence becomes logarithmic. It should be noted however, that the temperature of the arc was in the range of 1000–5000 K and probably cannot be compared fully to exposure to sunlight.
5. Conclusion
It has been shown that while significant research has been done on studying the degradation of thin polymer films there is still much to be done. In general, studies seem to have concentrated on specific applications. However, as thin polymer films become more important for the sustainability and development of new products, a more comprehensive approach must be sought in order to better understand degradation in general. In this way, polymer thin films can be optimised with a view of designing for the whole life cycle of products rather than remaining academic curiosities.References
- M. W. Keller, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2007, 17, 2399–2404 CrossRef CAS.
- M. W. Keller, S. R. White and N. R. Sottos, Polymer, 2008, 49, 3136–3145 CrossRef CAS.
- T. Kerle, Z. Lin, H.-C. Kim and T. P. Russell, Macromolecules, 2001, 34(10), 3484–3492 CrossRef CAS.
- J. G. Ibanez, A. Alatorre-Ordaz, S. Gutierrez-Granados and N. Batina, Polym. Degrad. Stab., 2008, 93, 827–837 CrossRef CAS.
- J. L. Keddie, R. A. L. Jones and R. A. Cory, Europhys. Lett., 1994, 27(1), 59–64 CrossRef CAS.
- J. H. van Zanten, W. E. Wallace and W. Wu, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1996, 53(3), 2053–2056 CrossRef.
- A. Torres, P. F. Nealy and J. J. de Pablo, Phys. Rev. Lett., 2000, 85(15), 3221–3224 CrossRef.
- F. D. Egitto and L. J. Matienzo, IBM J. Res. Dev., 1994, 38(4), 423–439 CrossRef CAS.
- Ullmann's Encyclopdeia of Industrial Chemistry, 6th edn, s.v. “Thin Films”, 2003 Search PubMed.
- Z. Nie and E. Kumacheva, Nat. Mater., 2008, 7, 277–290 CrossRef CAS.
- A. Hernandez-Santana and D. Graham, Nat. Nanotechnol., 2010, 5, 629–630 CrossRef CAS.
- X. Yao, T. Ito and D. A. Higgins, Langmuir, 2008, 24(16), 8939–8943 CrossRef CAS.
- M. Bompart, K. Haupt and C. Ayela, Top. Curr. Chem., 2012, 325, 83–110 CrossRef CAS.
- D. Li, C. Symonds, F. Bessueille, J. C. Plenet, A. Errachid, G. Wu, J. Shen and J. Bellessa, J. Opt. A: Pure Appl. Opt., 2009, 11, 065601 CrossRef.
- D. Cheyns, K. Vasseur, C. Rolin, J. Genoe, J. Poortmans and P. Heremans, Nanotechnology, 2008, 19, 424016 CrossRef CAS.
- K. Honda, M. Morita, H. Masunaga, S. Sasakai, M. Takata and A. Takahara, Soft Matter, 2010, 6, 870–875 RSC.
- J. Bowen, D. Cheneler and A. P. G. Robinson, Microelectron. Eng., 2012, 97, 34–37 CrossRef CAS.
- K. Salaita, Y. Wang and C. A. Mirkin, Nat. Nanotechnol., 2007, 2, 145–155 CrossRef CAS.
- S. W. Lee, R. G. Sanedrin, B.-K. Oh and C. A. Mirkin, Adv. Mater., 2005, 17(22), 2749–2753 CrossRef CAS.
- D. C. Coffey and D. S. Ginger, J. Am. Chem. Soc., 2005, 127(13), 4564–4565 CrossRef CAS.
- H. Jeon, R. Schmidt, J. E. Barton, D. J. Hwang, L. J. Gamble, D. G. Castner, C. P. Grigoropoulos and K. E. Healey, J. Am. Chem. Soc., 2011, 133(16), 6138–6141 CrossRef CAS.
- D. Credgington, O. Fenwick, A. Charas, J. Morgado, K. Suhling and F. Cacialli, Adv. Funct. Mater., 2010, 20(17), 2842–2847 CrossRef CAS.
- A. P. Kumar, D. Depan, N. S. Tomer and R. P. Singh, Prog. Polym. Sci., 2009, 34, 479–515 CrossRef CAS.
- S. Dowland, T. Lutz, A. Ward, S. P. King, A. Sudlow, M. S. Hill, K. C. Molloy and S. A. Haque, Adv. Mater., 2011, 23(24), 2739–2744 CrossRef CAS.
- M. Diepens and P. Gijsman, Polym. Degrad. Stab., 2009, 94(10), 1808–1813 CrossRef CAS.
- J. J. Senkevich, Chem. Vap. Deposition, 2011, 17(4–6), 170–172 CrossRef CAS.
- A. L. Holt, J. P. Bearinger, C. L. Evans and S. A. Carter, Sens. Actuators, B, 2010, 143(2), 600–605 CrossRef.
- T. Lippert, UV Laser Ablation of Polymers: From Structuring to Thin Film Deposition, in Laser-surface Interactions for New Materials Production, ed. A. Miotello and P. M. Ossi, Springer, Berlin, 2009 Search PubMed.
- S. De, T. M. Higgins, P. E. Lyons, E. M. Doherty, P. N. Mirmalra, W. J. Blau, J. J. Boland and J. N. Coleman, ACS Nano, 2009, 3(7), 1767–1774 CrossRef CAS.
- T. Miyasaka, J. Phys. Chem. Lett., 2011, 2(3), 262–269 CrossRef CAS.
- I. Armentano, M. Dottori, E. Fortunati, S. Mattiolo and J. M. Kenny, Polym. Degrad. Stab., 2010, 95, 2126–2146 CrossRef CAS.
- A. Jansson, K. Moller and T. Hjertberg, Polym. Degrad. Stab., 2004, 84, 227–232 CrossRef CAS.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41(12), 1578–1586 CrossRef CAS.
- D. Mordehai, S.-W. Lee, B. Backes, D. J. Srolovitz, W. D. Nix and E. Rabkin, Acta Mater., 2011, 59(13), 5202–5215 CrossRef CAS.
- K. P. Jain and M. A. El-Sayed, Chem. Phys. Lett., 2010, 487(4–6), 153–154 CrossRef.
- G. Walters and I. P. Parkin, J. Mater. Chem., 2009, 19, 574–590 RSC.
- J. Yang, E. Sargent, S. Kelley and J. Y. Ying, Nat. Mater., 2009, 8, 683–689 CrossRef CAS.
- M. Halik, H. Klauk, U. Zschieschang, G. Schmid, W. Radlik and W. Weber, Adv. Mater., 2002, 14(23), 1717–1722 CrossRef CAS.
- T. W. Kelley, D. V. Muyres, P. F. Baude, T. P. Smith and T. D. Jones, Mater. Res. Soc. Symp. Proc., 2003, 771, L.6.5.1 Search PubMed.
- S. Steudel, K. Myny, S. De Vusser, J. Genoe and P. Heremans, Appl. Phys. Lett., 2006, 89, 183503 CrossRef.
- J. H. Cho, J. Lee, Y. Xia, B. Kim, Y. He, M. J. Renn, T. P. Lodge and C. D. Frisbie, Nat. Mater., 2008, 7, 900–906 CrossRef CAS.
- C. Xu, H. Ho and S. A. Boggs, IEEE Electr. Insul. Mag., 2008, 24(6), 30–34 CrossRef.
- M. Goosey, Polym. Degrad. Stab., 1987, 17, 237–252 CrossRef CAS.
- D. C. Borrelli, M. C. Barr, V. Bulovic and K. K. Gleason, Sol. Energy Mater. Sol. Cells, 2012, 99, 190–196 CrossRef CAS.
- Z. Y. Hu, J. J. Zhang, S. Z. Xiong and Y. Zhao, Sol. Energy Mater. Sol. Cells, 2012, 99, 221–225 CrossRef CAS.
- M. I. Sabir, X. Xu and L. Li, J. Mater. Sci., 2009, 44, 5713–5724 CrossRef CAS.
- K. C. Shin, B. S. Kim, J. H. Kim, T. G. Park, J. D. Nam and D. S. Lee, Polymer, 2005, 46, 3801–3806 CrossRef CAS.
- P. F. McDonald, J. G. Lyons, L. M. Geever and C. L. Higginbotham, J. Mater. Sci., 2010, 45, 1284–1292 CrossRef CAS.
- N. Bege, S. O. Steinmuller, M. Kalinowski, R. Ruel, S. Klaus, H. Petersen, C. Curdy, J. Janek and T. Kissel, Eur. J. Pharm. Biopharm., 2012, 80(3), 562–570 CrossRef CAS.
- L. Xu and A. Yamamoto, Colloids Surf., B, 2012, 93, 67–74 CrossRef CAS.
- V. Gribova, R. Auzely-Velty and C. Picart, Chem. Mater., 2012, 24(5), 854–869 CrossRef CAS.
- X. J. Li, Q. J. Luo, Y. Huang, X. D. Li, F. Zhang and S. F. Zhao, Polym. Adv. Technol., 2012, 23(4), 756–764 CrossRef CAS.
- R. Zahn, E. Thomasson, O. Guillaume-Gentil, V. Janos and T. Zambelli, Biomaterials, 2012, 33(12), 3421–3427 CrossRef CAS.
- C. Borselli, H. Storrie, F. Benesch-Lee, D. Shvartsman, C. Cezar, J. W. Lichtman, H. H. Vandenburgh and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3287–3292 CrossRef CAS.
- H. Chu, J. Gao, C.-W. Chen, J. Huard and Y. Wang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13444–13449 CrossRef CAS.
- Z. Q. Zhang, C. H. Pan and D. Chung, Food Res. Int., 2011, 44(4), 1000–1007 CrossRef CAS.
- K. Hori, C. Sotozono, J. Hamuro, K. Yamasaki, Y. Kimura, M. Ozeki, Y. Tabata and S. Kinoshita, J. Controlled Release, 2007, 118, 169–176 CrossRef CAS.
- C. E. Mora-Huertas, H. Fessi and A. Elaissari, Int. J. Pharm., 2010, 385, 113–142 CrossRef CAS.
- K. Letchford and H. Burt, Eur. J. Pharm. Biopharm., 2007, 65(3), 259–269 CrossRef CAS.
- N. Anton, J. P. Benoit and P. Saulnier, J. Controlled Release, 2008, 128(3), 185–199 CrossRef CAS.
- S. Zhao, C. Y. Hu, X. Y. Chen, J. Zhou, Y. H. Jiao, K. Zhang and Y. Fu, Soft Matter, 2012, 8(4), 937–941 RSC.
- Z. D. Kihm, E. M. Veen, J. D. Bergen-Hartigan, Y. Zhang and Y. Liu, Anal. Sci., 2012, 28(2), 183–186 CrossRef CAS.
- W. Zhang, S. Lin, C. Wang, J. Hu, C. Li, Z. Zhuang, Y. Zhou, R. A. Mathies and J. Chaoyong Yang, Lab Chip, 2009, 9, 3088–3094 RSC.
- P. Kumlangdudsana, A. Tuantranont, S. T. Dubas and L. Dubas, Mater. Lett., 2011, 65(23–24), 3629–3632 CrossRef CAS.
- D. Briassoulis, Polym. Degrad. Stab., 2007, 92, 1115–1132 CrossRef CAS.
- S. Beckford, N. Langston, M. Zou and R. Wei, Appl. Surf. Sci., 2011, 257, 5688–5693 CrossRef CAS.
- T. Caykara and O. Guven, Polym. Degrad. Stab., 1999, 65, 225–229 CrossRef CAS.
- D. Seghete, B. D. Davidson, R. A. Hall, Y. J. Chang, V. M. Bright and S. M. George, Sens. Actuators, A, 2009, 155(1), 8–15 CrossRef.
- P. Alonso-Magdalena, S. Morimoto, C. Ripoll, E. Fuentes and A. Nadal, Environ. Health Perspect., 2006, 114(1), 106–112 CrossRef CAS.
- C. Z. Yang, S. I. Yaniger, V. C. Jordan, D. J. Klein and G. D. Bittner, Environ. Health Perspect., 2011, 119(7), 989–996 CrossRef CAS.
- Thomson Reuters (n.d.), Web of Knowledge, www.wok.mimas.ac.uk/, accessed 16 March 2012 Search PubMed.
- R. G. Bayer, P. A. Engel and E. Sacher, Wear, 1975, 32, 181–194 CrossRef CAS.
- S. K. Venkataraman, J. C. Nelson, A. J. Hsieh, D. L. Kohlstedt and W. W. Gerberich, J. Adhes. Sci. Technol., 1993, 7(12), 1279–1292 CrossRef CAS.
- B. D. Ratner, K. W. Gladhill and T. A. Horbett, J. Biomed. Mater. Res., 1988, 22(6), 509–527 CrossRef CAS.
- J. P. Collier, M. B. Mayor, J. L. McNamara, V. A. Surprenant and R. E. Jensen, Clin. Orthop. Relat. Res., 1991, 273, 232–242 Search PubMed.
- S. Tagawa, Adv. Polym. Sci., 1993, 105, 99–116 CrossRef CAS.
- L. Eggbert, J. Friedridh, H. Wittrich, M. Vonlowis and W. Abraham, Acta Polym., 1990, 41(11), 569–574 CrossRef.
- D. Latev, V. Dorfman and B. Pypkin, Surf. Coat. Technol., 1991, 47(1–3), 308–314 CrossRef CAS.
- R. Koksbang, I. I. Olsen, P. E. Tonder, N. Knudsen and D. Fauteux, J. Appl. Electrochem., 1991, 21(4), 301–307 CrossRef CAS.
- K. Itaya and I. Uchida, Membrane, 1986, 11(2), 83–91 CrossRef CAS.
- G. Jobst, G. Urbam, J. A. Jachimowicz, F. Kohl, O. Tilado, I. Lettenbichler and G. Nauer, Biosens. Bioelectron., 1993, 8(3–4), 123–128 CrossRef CAS.
- T. Lippert, Plasma Processes Polym., 2005, 2, 525–546 CrossRef CAS.
- H. Yasuda, Luminous Chemical Vapor Deposition and Interface Engineering, CRC Press, 2004 Search PubMed.
- A. Dasari, Z.-Z. Yu and Y.-W. Mai, Mater. Sci. Eng., R, 2009, 63, 31–80 CrossRef.
- P. V. Pavoor, B. P. Gearing, O. Muratoglu, R. E. Cohen and A. Bellare, Biomaterials, 2004, 27(8), 1527–1533 CrossRef.
- R. J. Varley, E. K. Simmonds, J. E. Seebergh and D. H. Berry, Prog. Org. Coat., 2011, 74(4), 679–686 CrossRef.
- N. K. Myshkin, M. I. Petrokovets and A. V. Kovalev, Tribol. Int., 2005, 38(11–12), 910–921 CrossRef CAS.
- S. S. Pesetskii, S. P. Bogdanovich and N. K. Myshkin, Trib. Polym. Nanocomposites, 2008, 55, 82–107 CrossRef.
- B. Singh and N. Sharma, Polym. Degrad. Stab., 2008, 93, 561–584 CrossRef CAS.
- M. Conroy, Wear, 2009, 266, 502–506 CrossRef CAS.
- M. Kappes, G. S. Frankel and N. Sridhar, Prog. Org. Coat., 2010, 60, 57–72 CrossRef.
- M. Rutkowska, K. Krasowska, A. Heimowska, I. Steinka and H. Janik, Polym. Degrad. Stab., 2002, 76(2), 233–239 CrossRef CAS.
- T. Artham and M. Doble, J. Polym. Environ., 2009, 17, 170–180 CrossRef CAS.
- P. C. Hiemenz, Polymer Chemistry: The Basic Concepts, Marcel Dekker, Inc., New York, USA, 1984 Search PubMed.
- J.-M. Lehn, Polym. Int., 2002, 51(10), 825–839 CrossRef CAS.
- S. Carraher, Polymer Chemistry, Marcel Dekker, Inc., New York, USA, 6th edn, 2003 Search PubMed.
- K. J. Saunders, Organic Polymer Chemistry: An Introduction to the Organic Chemistry of Adhesives, Fibres, Paints, Plastics and Rubbers, Chapman and Hall Ltd., London, UK, 2nd edn, 1988 Search PubMed.
- W. E. Driver, Plastics Chemistry and Technology, Van Nostrand Reinhold, New York, USA, 1979 Search PubMed.
- K. Klaus, Chemical Reactions in Plastics Processing, Hanser Publishers, New York, USA, 1987 Search PubMed.
- V. W. Y. Chio, C. K. M. Ng, D. Nikezic, T. Konishi and K. N. Yu, Nucl. Instrum. Methods Phys. Res., Sect. A, 2011, 631(1), 6–11 CrossRef.
- H. Park, S. Chio, J.-P. Lee and S. Park, J. Mater. Chem., 2011, 21, 11996–12000 RSC.
- D. Cheneler, J. Bowen, S. D. Evans, M. Gorzny, M. J. Adams and M. C. L. Ward, Polym. Degrad. Stab., 2011, 96(4), 561–565 CrossRef CAS.
- P. S. Ho, J. Vac. Sci. Technol., A, 1985, 3(3), 739–745 CAS.
- P. Engel, IEEE Trans. Reliab., 1993, 42(2), 262–267 CrossRef.
- H. Bohm, S. Betz and A. Ball, Tribol. Int., 1990, 23(6), 399–406 CrossRef CAS.
- K. Kato, Wear, 1992, 153(1), 277–295 CrossRef CAS.
- J. Bijwe, J. Indumathi, J. J. Rajesh and M. Fahim, Wear, 2001, 249(8), 715–726 CrossRef CAS.
- M. R. Chitsaz-Zadeh and N. S. Eiss Jr, Wear, 1986, 110(3–4), 359–368 CrossRef CAS.
- S. K. Biswas and K. Vijayan, Wear, 1992, 158, 193–211 CrossRef CAS.
- S. Iwamori, N. Hasegawa, A. Uemura, T. Tanabe and I. Nishiyama, Vacuum, 2010, 84, 592–596 CrossRef.
- N. Satyanarayana, S. K. Sinha and S.-C. Lim, J. Mater. Res., 2009, 24(11), 3331–3337 CrossRef CAS.
- S. M. Kurtz, O. K. Muratoglu, M. Evans and A. A. Edidin, Biomaterials, 1999, 20, 1659–1688 CrossRef CAS.
- P. V. Pavoor, B. P. Gearing, A. Bellare and R. E. Cohen, Wear, 2004, 256, 1196–1207 CrossRef CAS.
- M. D. Kriese, D. A. Boismier, N. R. Moody and W. W. Gerberich, Eng. Fract. Mech., 1998, 61, 1–20 CrossRef.
- B. Li, S.-Q. Huang and X.-Q. Feng, Arch. Appl. Mech., 2010, 80(2), 175–188 CrossRef.
- T. Q. Lu, W. X. Zhang and T. J. Wang, Int. J. Eng. Sci., 2011, 49, 967–975 CrossRef.
- B. Li, Y.-P. Cao, X.-Q. Feng and H. Gao, Soft Matter, 2012, 8, 5728–5745 RSC.
- D. Y. Liu and W. Q. Chen, Mech. Res. Commun., 2010, 37, 520–524 CrossRef.
- P. M. Reis, Nat. Mater., 2011, 10, 907–909 CrossRef CAS.
- J. Y. Chung, A. J. Nolte and C. M. Stafford, Adv. Mater., 2011, 23, 349–368 CrossRef CAS.
- V. A. Bely, A. I. Sviridenok, M. I. Petrokovets and V. G. Savkin, Friction and Wear in Polymer-based Materials, Pergamon Press, Oxford, 1982 Search PubMed.
- M. Godet, Wear, 1984, 100, 437–452 CrossRef.
- J. K. Pandey, R. K. Reddy, P. A. Kumar and R. P. Singh, Polym. Degrad. Stab., 2005, 88, 234–250 CrossRef CAS.
- G. V. Vinogradov, G. M. Bartenev, A. I. Elkin and V. K. Mikhaylov, Wear, 1970, 16, 213–219 CrossRef CAS.
- Y. Kojima, A. Usuki, M. Kawasumi, A. Okada, T. Kurauchi and O. Kamigaito, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 983–986 CrossRef CAS.
- S. S. Ray and M. Okamoto, Prog. Polym. Sci., 2003, 28, 1539–1641 CrossRef CAS.
- A. Dasari, Z.-Z. Yu and Y.-W. Mai, Trib. Polym. Nanocomposites, 2008, 55, 374–399 CrossRef.
- J.-Y. Lee, D.-P. Lim and D.-S. Lim, Composites, Part B, 2007, 38, 810–816 CrossRef.
- D. L. Burris, B. Boesl, G. R. Bourne and W. G. Sawyer, Macromol. Mater. Eng., 2007, 292, 387–402 CrossRef CAS.
- R. Domke and M. Radmacher, Langmuir, 1998, 14, 3320–3325 CrossRef.
- B. Poon, D. Rittel and G. Ravichandran, Int. J. Solids Struct., 2008, 45(24), 6018–6033 CrossRef.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 2004, 19(1), 3–20 CrossRef CAS.
- A. C. Fischer-Cripps, Nanoindentation, Springer-Verlag, NY, 2nd edn, 2004 Search PubMed.
- E. K. Dimitriadis, F. Horkay, J. Maresca, B. Kacha and R. S. Chadwick, Biophys. J., 2002, 82, 2798–2810 CrossRef CAS.
- I. N. Sneddon, Proc. Cambridge Philos. Soc., 1948, 44(4), 492–507 CrossRef.
- I. N. Sneddon, Int. J. Eng. Sci., 1965, 3, 47–57 CrossRef.
- T. S. Wu and Y. P. Chiu, Q. Appl. Math., 1967, 25, 233–240 Search PubMed.
- R. Dhaliwal, Int. J. Eng. Sci., 1970, 8, 273–288 CrossRef.
- R. Dhaliwal and I. Rau, Int. J. Eng. Sci., 1970, 8, 843–856 CrossRef.
- W. T. Chen and P. A. Engel, Int. J. Solids Struct., 1972, 8, 1257–1263 CrossRef.
- H. Y. Yu, S. C. Sanday and B. B. Rath, J. Mech. Phys. Solids, 1989, 38, 745–764 CrossRef.
- X. Chen and J. J. Vlassak, J. Mater. Res., 2001, 16, 2974–2982 CrossRef CAS.
- Y. T. Cheng and C. M. Cheng, Mater. Sci. Eng., 2004, 44, 91–149 CrossRef.
- D. Lebouvier, P. Gilormini and E. Felder, Thin Solid Films, 1989, 172, 227–239 CrossRef CAS.
- D. Lebouvier, P. Gilormini and E. Felder, J. Phys. D: Appl. Phys., 1985, 18, 199–210 CrossRef.
- X. Chen, N. Ogasawara, M. Zhao and N. Chiba, J. Mech. Phys. Solids, 2007, 55, 1618–1660 CrossRef.
- J. L. Bucaille, S. Stauss, E. Felder and J. Michler, Acta Mater., 2003, 51, 1663–1678 CrossRef CAS.
- Y. P. Cao and J. Lu, Acta Mater., 2004, 52, 1143–1153 CrossRef CAS.
- O. Casals and J. Alcala, Acta Mater., 2005, 53, 3545–3561 CrossRef CAS.
- Y. Lu and D. M. Shinozaki, J. Mater. Sci., 2002, 37, 1283–1293 CrossRef CAS.
- S. Sills, H. Fong, C. Buenviaje, M. Sarikaya and R. M. Overney, J. Appl. Phys., 2005, 98, 014302 CrossRef.
- M. V. Swain and J. Mencik, Thin Solid Films, 1994, 253, 204–211 CrossRef CAS.
- G. M. Odegard, T. Bandorawalla, H. M. Herring and T. S. Gates, 2003 SEM Annual Conference and Exposition on Experimental and Applied Mechanics, Charlotte, NC, 2003 Search PubMed.
- Y. Yin, S.-F. Ling and Y. Liu, Mater. Sci. Eng., A, 2004, 379, 334–340 CrossRef.
- M. Vandamme and F. J. Ulm, Int. J. Solids Struct., 2006, 43, 3142–3165 CrossRef.
- L. Cheng, X. Xia, L. E. Scriven and W. W. Gerberich, Mech. Mater., 2005, 37, 213–226 CrossRef.
- M. L. Oyen, Philos. Mag., 2006, 86, 1–17 CrossRef.
- J. R. M. Radok, Q. Appl. Math., 1957, 15, 198–202 Search PubMed.
- E. H. Lee and J. R. M. Radok, J. Appl. Mech., 1960, 27, 438–444 CrossRef.
- S. C. Hunter, J. Mech. Phys. Solids, 1960, 8, 219–234 CrossRef.
- T. C. T. Ting, J. Appl. Mech., 1966, 88, 845–854 CrossRef.
- E. M. Darling, S. Zauscher and F. Guilak, Osteoarthritis Cartilage, 2006, 14(6), 571–579 CrossRef CAS.
- J. F. Archard, Proc. R. Soc. London, Ser. A, 1957, 243, 190–205 CrossRef.
- J. F. Archard and M. T. Kirk, Proc. R. Soc. London, Ser. A, 1961, 261, 532–550 CrossRef.
- J. F. Archard, J. Appl. Phys., 1953, 24, 981–989 CrossRef.
- J. F. Archard and W. Hirst, Proc. R. Soc. London, Ser. A, 1957, 238, 515–528 CrossRef CAS.
- G. M. Hamilton and L. E. Goodman, J. Appl. Mech., 1966, 33, 371–376 CrossRef.
- E. So, M. C. Demirel and K. J. Wahl, J. Phys. D: Appl. Phys., 2010, 43, 045403 CrossRef.
- J. Lintymer, N. Martin, J.-M. Chappé, J. Takadoum and P. Delobelle, Thin Solid Films, 2006, 503, 177–189 CrossRef CAS.
- A. Y. Cao, P. L. Dickrell, W. G. Sawyer, M. N. Ghasemi-Nejhad and P. M. Ajayan, Science, 2005, 310, 1307–1310 CrossRef CAS.
- F. Wredenberg and P.-L. Larsson, J. Coat. Technol. Res., 2010, 7, 623–635 CrossRef CAS.
- P. L. Larsson and F. Wredenberg, J. Phys. D: Appl. Phys., 2008, 41, 074022 CrossRef.
- F. Wredenberg and P. L. Larsson, J. Mech. Mater. Struct., 2009, 4, 1041–1062 CrossRef.
- F. Wredenberg and P. L. Larsson, J. Coat. Technol. Res., 2010, 7, 279–290 CrossRef CAS.
- R. M. Overney, E. Meyer, J. Frommer, H.-J. Guentherodt, M. Fujihira, H. Takano and Y. Gotoh, Langmuir, 1994, 10(4), 1281–1286 CrossRef CAS.
- B. J. Briscoe and S. K. Sinha, Materialwiss. Werkstofftech., 2003, 34(10–11), 989–1002 CrossRef CAS.
- M. Wang, K. M. Liechti, J. M. White and R. M. Winter, J. Mech. Phys. Solids, 2004, 52, 2329–2354 CrossRef CAS.
- N. Satyanarayana, S. K. Sinha and S. L. Shen, Tribol. Lett., 2007, 28, 71–80 CrossRef CAS.
- B. J. Briscoe and D. Tabor, J. Adhes., 1978, 9, 145–155 CrossRef CAS.
- B. Du, M. R. Van Landingham, Q. Zhang and T. He, J. Mater. Res., 2001, 16, 1487–1482 CrossRef CAS.
- P. Y. Chiu, J. R. Vail, P. R. Barry, S. S. Perry, S. R. Phillpot, W. G. Sawyer and S. B. Sinnott, Tribol. Lett., 2012, 45(5), 79–87 CrossRef CAS.
- M. M. Yanadi, S. G. Soetanto and S. K. Sinha, Tribol. Lett., 2011, 41(1), 217–226 CrossRef.
- F. D. Egitto and L. J. Matienzo, IBM J. Res. Dev., 1994, 38, 423–439 CrossRef CAS.
- C. A. L. Westerdahl, J. R. Hall, E. C. Schramm and D. W. Levi, J. Colloid Interface Sci., 1974, 47, 610–620 CrossRef CAS.
- M. Reiche, U. Gosele and M. Wiegand, Cryst. Res. Technol., 2000, 35, 807–821 CrossRef CAS.
- C.-M. Chan, T.-M. Ko and H. Hiraoka, Surf. Sci. Rep., 1996, 24, 1–54 CrossRef CAS.
- R. H. Hansen, J. V. Pascale, T. De Benedictis and P. M. Rentzepis, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 2205–2214 CrossRef CAS.
- J. G. Ibanez, A. Alatorre-Ordaz, S. Gutierrez-Granados and N. Batina, Polym. Degrad. Stab., 2008, 93, 827–837 CrossRef CAS.
- S. Steudel, K. Myny, S. De Vusser, J. Genoe and P. Heremans, Appl. Phys. Lett., 2006, 89, 183503 CrossRef.
- L. Zhou, S. Park, B. Bai, J. Sun, S.-C. Wu, T. N. Jackson, S. Nelson, D. Freeman and Y. Hong, IEEE Electron Device Lett., 2005, 26, 640–642 CrossRef CAS.
- N. J. H. Al-Mashhadan and S. Mohamad, J. Eng. Tech., 2011, 29(1), 20–32 Search PubMed.
- T. Caykara and O. Guven, Polym. Degrad. Stab., 1999, 65, 225–229 CrossRef CAS.
- J. M. Taguenang, A. Kassu, P. B. Ruffin, C. Brantley, E. Edwards and A. Sharma, Opt. Commun., 2008, 281, 2089–2092 CrossRef CAS.
- B. Littlejohn, K. M. Heeger, T. Wise, E. Gettrust and M. Lyman, J. Instrum., 2009, 4, T09001 CrossRef.
- C. D. Easton and M. V. Jacob, Polym. Degrad. Stab., 2009, 94, 597–609 CrossRef CAS.
- T. R. Gengenbach and H. J. Griesser, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 985–1000 CrossRef CAS.
- T. R. Gengenbach and H. J. Griesser, Polymer, 1999, 40, 5079–5094 CrossRef CAS.
- D. Briassoulis, Polym. Degrad. Stab., 2005, 88, 485–503 CrossRef.
- A. Jansson, K. Moller and T. Hjertberg, Polym. Degrad. Stab., 2004, 84, 227–232 CrossRef CAS.
- M. Jørgensen, K. Norrman and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92, 686–714 CrossRef.
- M.-D. Damaceanu, R.-D. Rusu, M. Olaru and M. Bruma, Proc. IEEE, 2010, 231–234 CAS.
- H. Jeon, R. Schmidt, J. E. Barton, D. J. Hwang, L. J. Gamble, D. G. Castner, C. P. Grigoropoulos and K. E. Healy, J. Am. Chem. Soc., 2011, 133, 6138–6141 CrossRef CAS.
- R. J. Lade, I. W. Morley, P. W. May, K. N. Rosser and M. N. R. Ashfold, Diamond Relat. Mater., 1999, 8, 1654–1658 CrossRef CAS.
- G. B. Blanchet, C. R. Fincher Jr, C. L. Jackson, S. I. Shah and K. H. Gardner, Science, 1993, 292, 719–721 Search PubMed.
- A. Palla-Papavlu, V. Dinca, V. Ion, A. Moldovan, B. Mitu, C. Luculescu and M. Dinescu, Appl. Surf. Sci., 2011, 257(12), 5303–5307 CrossRef CAS.
- L. Urech, T. Lippert, C. R. Phipps and A. Wokaun, Appl. Surf. Sci., 2007, 253(15), 6409–6415 CrossRef CAS.
- H. Sato, J. Photochem. Photobiol., C, 2001, 2, 139–152 CrossRef CAS.
- E. Rebollar, G. Bounos, M. Oujja, C. Domingo, S. Georgiou and M. Castillejo, Appl. Surf. Sci., 2005, 248(1–4), 254–258 CrossRef CAS.
- N. Arnold and N. Bityurin, Appl. Phys. A, 1999, 68, 615–625 CrossRef CAS.
- N. Bityurin, B. S. Lukyanchuk, M. H. Hong and T. C. Chong, Chem. Rev., 2003, 103, 519–552 CrossRef CAS.
- J. F. Rabek, Mechanism of Photophysical Processes and Photochemical Reactions in Polymers, John Wiley & Sons, 1987 Search PubMed.
- W. Navarrini, M. V. Diamanti, M. Sansotera, F. Persico, W. Menghua, L. Magagnin and S. Radice, Prog. Org. Coat., 2012, 74(4), 794–800 CrossRef CAS.
- K. I. Priyadarsini, J. Photochem. Photobiol., C, 2009, 10(2), 81–95 CrossRef CAS.
- W. R. Browne, Coord. Chem. Rev., 2008, 252, 2470–2479 CrossRef CAS.
- S. Liu, S. Kokot and G. Will, J. Photochem. Photobiol., C, 2009, 10, 159–172 CrossRef CAS.
- C. Wochnowski, M. A. S. Eldin and S. Metev, Polym. Degrad. Stab., 2005, 89, 252–264 CrossRef CAS.
- X. Xingsheng, M. Hai, Z. Qijing and Z. Yunsheng, J. Opt. A: Pure Appl. Opt., 2002, 4, 237–242 CrossRef.
- S. Kuper and M. Stuke, Appl. Phys. A: Solids Surf., 1989, 49, 211–215 CrossRef.
- V. V. Nossov, B. Hage, B. Jusselin and C. Flevet, Tech. Phys., 2007, 77, 117–126 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |