DOI:
10.1039/C3SC52799A
(Edge Article)
Chem. Sci., 2014,
5, 942-950
Reactivity of uranium(IV) bridged chalcogenido complexes UIV–E–UIV (E = S, Se) with elemental sulfur and selenium: synthesis of polychalcogenido-bridged uranium complexes†
Received
8th October 2013
, Accepted 7th November 2013
First published on 8th November 2013
Abstract
We report the syntheses, electronic properties, and molecular structures of a series of polychalcogenido-bridged dinuclear uranium species. These complexes are supported by the sterically encumbering but highly flexible, single N-anchored tris(aryloxide) chelator (AdArO)3N3−. Reaction of an appropriate uranium precursor, either the U(III) starting material, [((AdArO)3N)U(DME)], or the dinuclear mono-chalcogenido-bridged uranium(IV/IV) compounds [{((AdArO)3N)U(DME)}2(μ-E)] (E = S, Se), with elemental sulfur or selenium, yields new complexes with a variety of bridging chalcogenide entities μ-Emn− (E = S, m = 2, n = 1 or 2 and E = Se, m = 2, 4; n = 2). Activation of the heavy chalcogens typically requires either a coordinatively unsaturated, strongly-reducing metal complex or a compound with a metal–metal bond. Since uranium complexes in the +IV oxidation state, are generally considered rather unreactive, the observed reaction of the here employed uranium(IV)/(IV) species with elemental chalcogens is fairly remarkable.
Introduction
Actinide chalcogenido complexes containing the group 16 congeners sulfur, selenium, and tellurium have been known for three decades.1–13 However, only recently elemental chalcogen activation and chalcogen atom transfer reactions with actinide complexes have been developed.14–22 This renewed interest in actinide reactivity with the chalcogens and chalcogenides can – to some extent – be attributed to the importance of solid-state materials that are obtained from controlled pyrolysis of inorganic or organometallic chalcogenolates.23 This family of compounds holds promise for various applications, including photovoltaics,24 fast-ion conductors,25,26 and thermoelectric energy conversion,27–29 among others.30–33 From a basic research point of view, complexes containing the hard uranium ion and soft sulfur, selenium, and tellurium ligands are fundamentally important to advance understanding of covalency in the metal ligand bond that is essential for the development of lanthanide/actinide separation methodologies.34–36
In uranium coordination chemistry, chalcogenido ligands tend to prefer bridging coordination modes.6–9,14,19,37 However, by utilizing an ylide-masked U(III) starting material that slows the rate of comproportionation, and thus prevents the formation of bridging chalcogenide complexes, the monomeric, terminal chalcogenide species [H3CPPh3][(R2N)3U(E)] (E = S, Se, Te; R = SiMe3) were synthesized recently by Hayton et al.38
Surprisingly, despite the propensity of the heavier chalcogenides to catenate to rings and chains of various sizes,32,33 there are only a few actinide complexes featuring polychalcogenide ligands.
The few existing complexes include the thorium pentasulfido complex [Cp*2ThS5], that was obtained via salt metathesis reaction of [Cp*2ThCl2] with Na2S5 by Sattelberger et al.,2 and the homoleptic diselenido complex K4[U(Se2)4], synthesized by Kanatzidis et al. via molten salt synthetic techniques.39 Additionally, Boncella et al. were able to synthesize the two tetrachalcogenido complexes [{(tBu2bpy)U(NtBu)2(I)}2(μ-η2:η2-E4)] (E = S, Se) by activating elemental sulfur and selenium, respectively, with a dinuclear uranium(V/V) halide complex that serves as a two-electron reductant.37
Herein, we report the syntheses, electronic properties, and molecular structures of a series of dinuclear uranium polychalcogenido complexes of the type [{((AdArO)3N)UIV}2(μ-Emn−)] with μ-Emn− = S, m = 2, n = 1 or 2, and E = Se, m = 2, 4; n = 2. These new compounds can be synthesized in a remarkably controlled manner just by the appropriate choice of uranium precursor and the stoichiometric addition of the elemental chalcogen.
Results and discussion
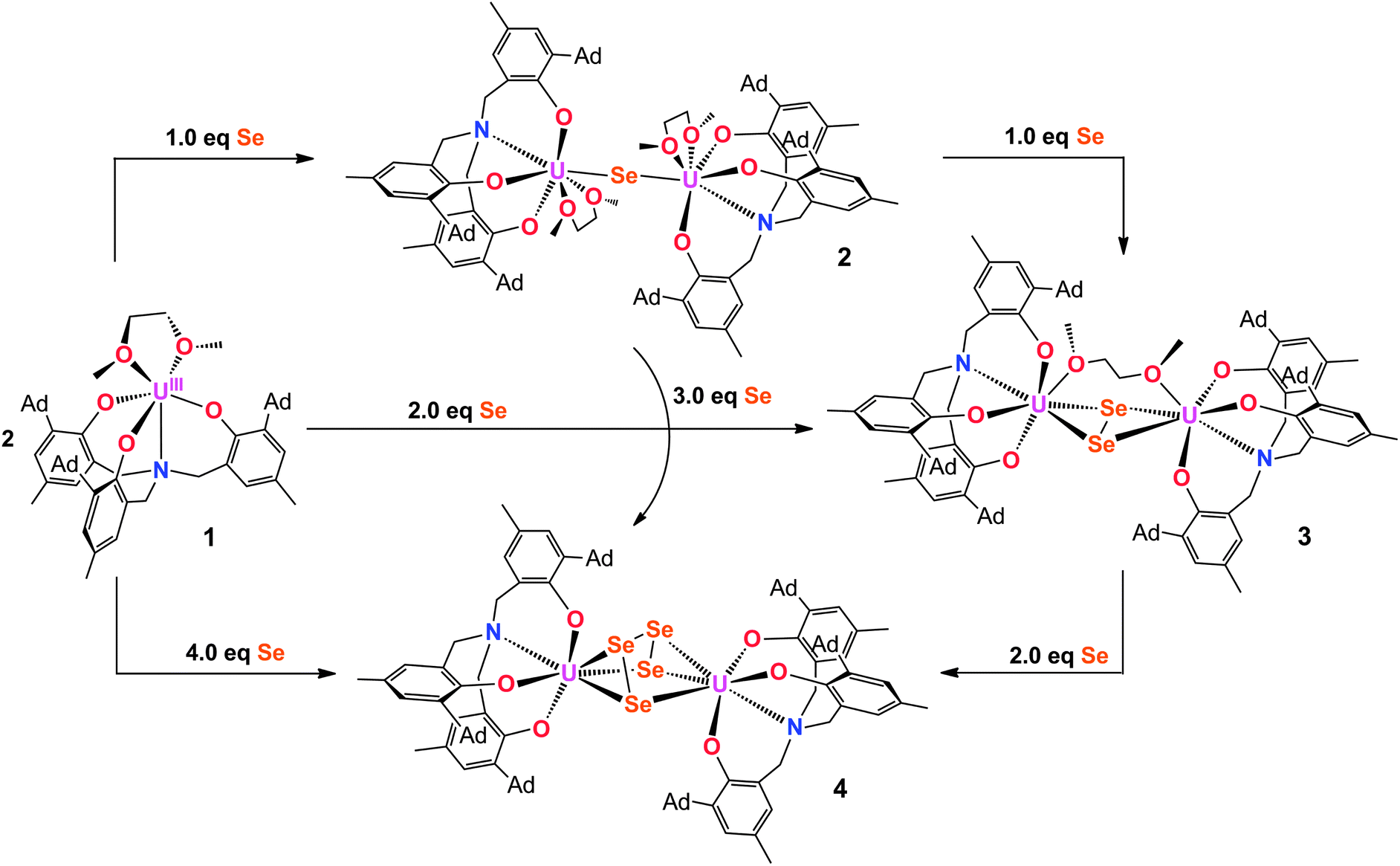
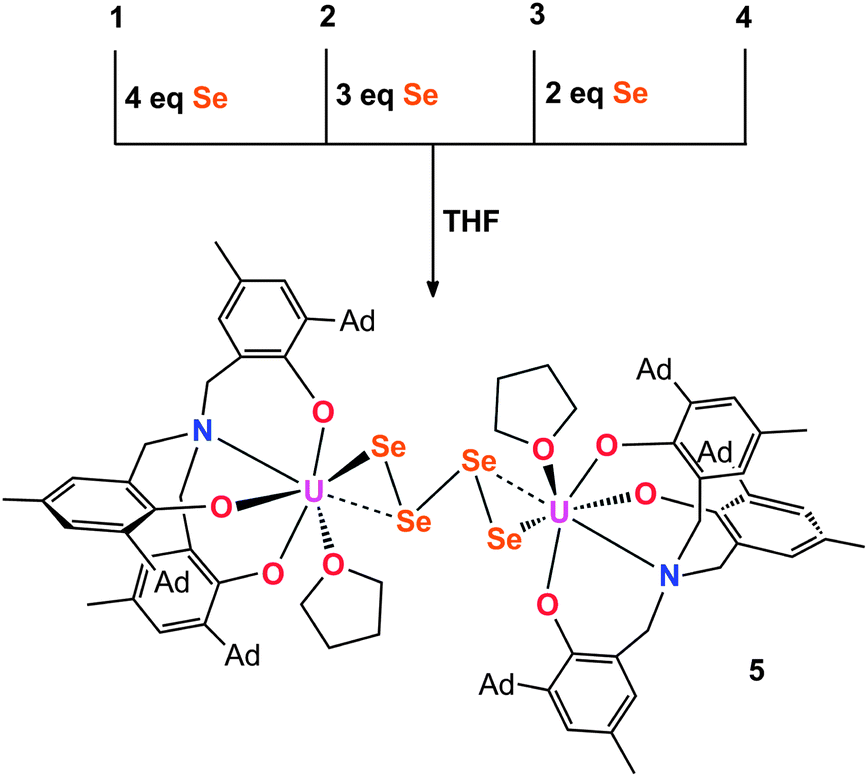
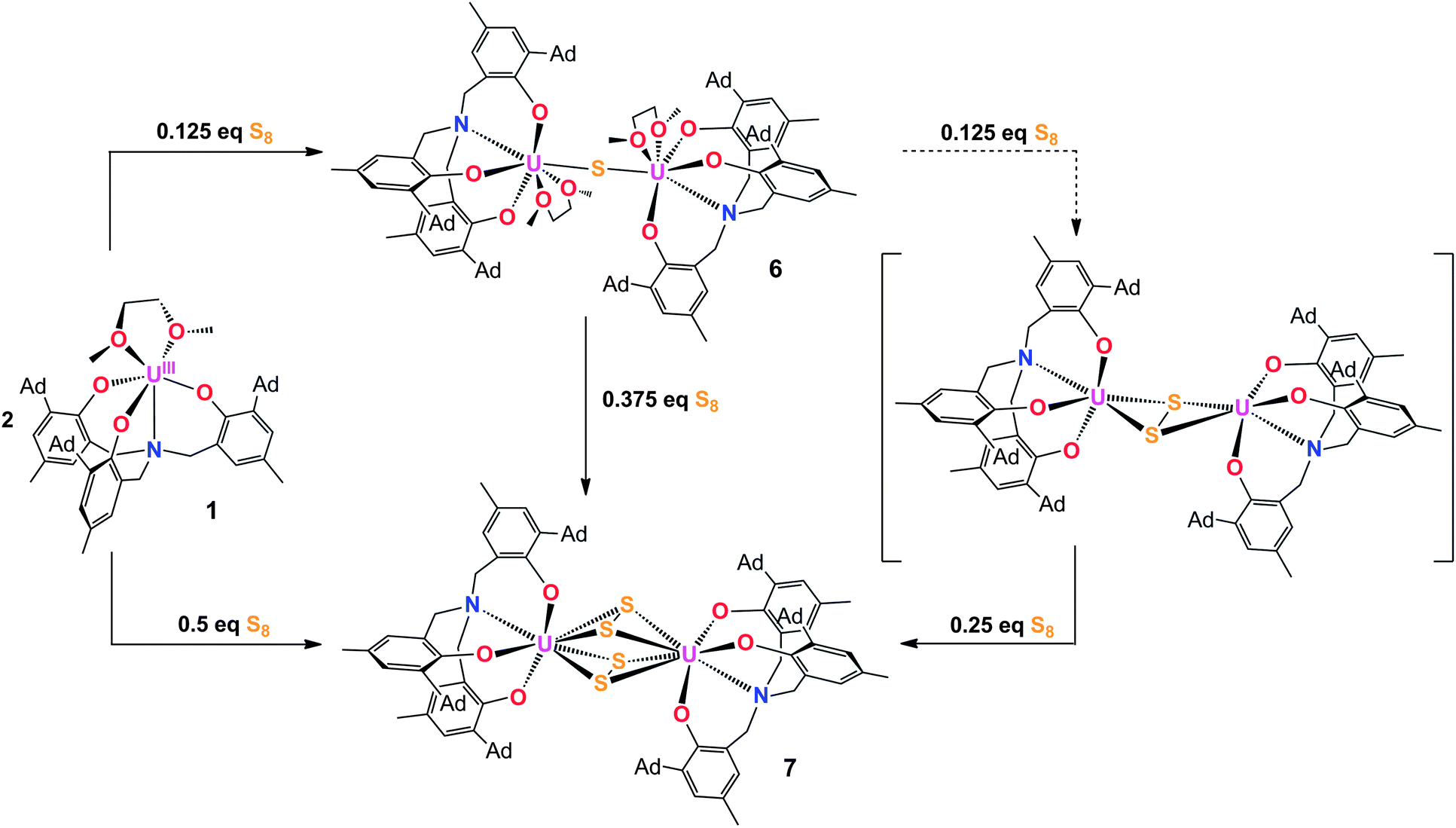
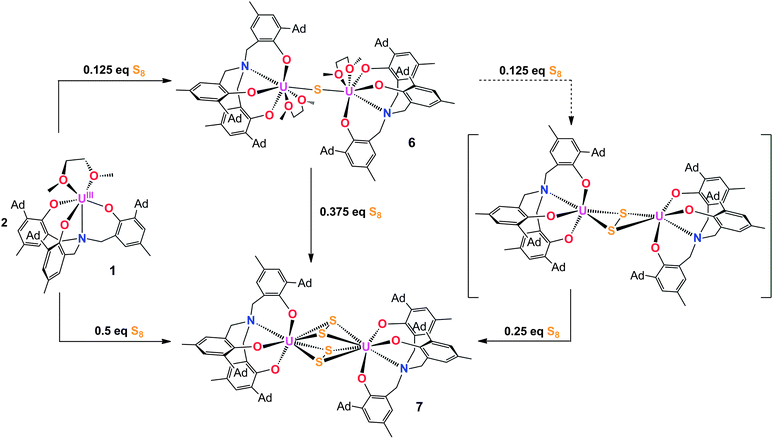
Employing the tacn (tacn = 1,4,7-triazacyclononane) and single N-anchored tris(aryloxide) ligands, we recently reported a series of chalcogen-bridged compounds containing purely inorganic chalcogenido ligands, [{((tBu,tBuArO)3tacn)UIV}2(μ-E)] and [{((AdArO)3N)UIV(DME)}2(μ-E)] (E = S, Se), that can be synthesized from the uranium(III) starting materials [((tBu,tBuArO)3tacn)UIII] and [((AdArO)3N)UIII(DME)] with stoichiometric amounts of elemental sulfur or selenium, respectively.14 The sterically shielded but potentially reactive μ-E bridging sulfido- and selenido-ligands in complexes of the (tBu,tBuArO)3tacn3− chelator [{((tBu,tBuArO)3tacn)UIV}2(μ-E)] (E = S, Se) do not undergo any further reactions with the elemental chalcogens or, e.g., CO2. Likewise, the complex of the bridging oxygen analogue [{((tBu,tBuArO)3tacn)UIV}2(μ-O)] does not exhibit such reactivity. In contrast, we could show that complexes of the N-anchored ligand system, [{((AdArO)3N)UIV(DME)}2(μ-E)] (E = O, S, Se), easily react with CO2 and even the heterocumulene analogues CS2 and COS to form dinuclear U(IV/IV) mixed-carbonate complexes.20,21,40 This observation is attributed to the higher flexibility of the tetradentate (AdArO)3N3− ligand compared to the hexadentate (tBu,tBuArO)3tacn3− chelate. To further probe the reactivity of [{((AdArO)3N)UIV(DME)}2(μ-Se)] (2) and [{((AdArO)3N)UIV(DME)}2(μ-S)] (6), we thoroughly investigated its proclivity to react with chalcogens by applying precise stoichiometric amounts of elemental sulfur and selenium. Tellurium did not show any reactivity; nor is the corresponding Te-bridged precursor [{((AdArO)3N)UIV(DME)}2(μ-Te)] accessible. However, applying this method to the U(III) precursor and mono S- and Se-bridged U(IV/IV) species, we were able to synthesize compounds [{((AdArO)3N)UIV}2(μ-Se2)(μ-DME)] (3), [{((AdArO)3N)UIV}2(μ-η3:η3-Se4)] (4), [{((AdArO)3N)UIV(THF)}2(μ-η2:η2-Se4)] (5), and [{((AdArO)3N)U}2(μ-S2)2] (7) that feature a variety of different di- and tetra-chalcogenido bridging ligands (Chart 1). These bridging polychalcogenido uranium complexes are a rare species with only few examples in actinide chemistry.2,37,39 Interestingly, Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E (E = S, Se), known to be a potent chalcogen atom transfer reagent, did not react with the bridging chalcogenido ligands of complexes 2 and 6.41 A series of mixed-chalcogenido bridged complexes can be obtained by reacting selenium bridged complex 2, or sulfur bridged complex 6, with precisely one equiv. of elemental sulfur or selenium, respectively. Due to the expected strong disorder in the resulting single crystals we did not further pursue the isolation of these compounds.
E (E = S, Se), known to be a potent chalcogen atom transfer reagent, did not react with the bridging chalcogenido ligands of complexes 2 and 6.41 A series of mixed-chalcogenido bridged complexes can be obtained by reacting selenium bridged complex 2, or sulfur bridged complex 6, with precisely one equiv. of elemental sulfur or selenium, respectively. Due to the expected strong disorder in the resulting single crystals we did not further pursue the isolation of these compounds.
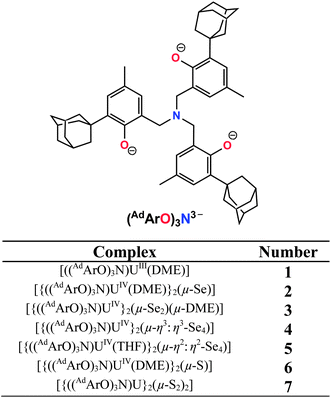 |
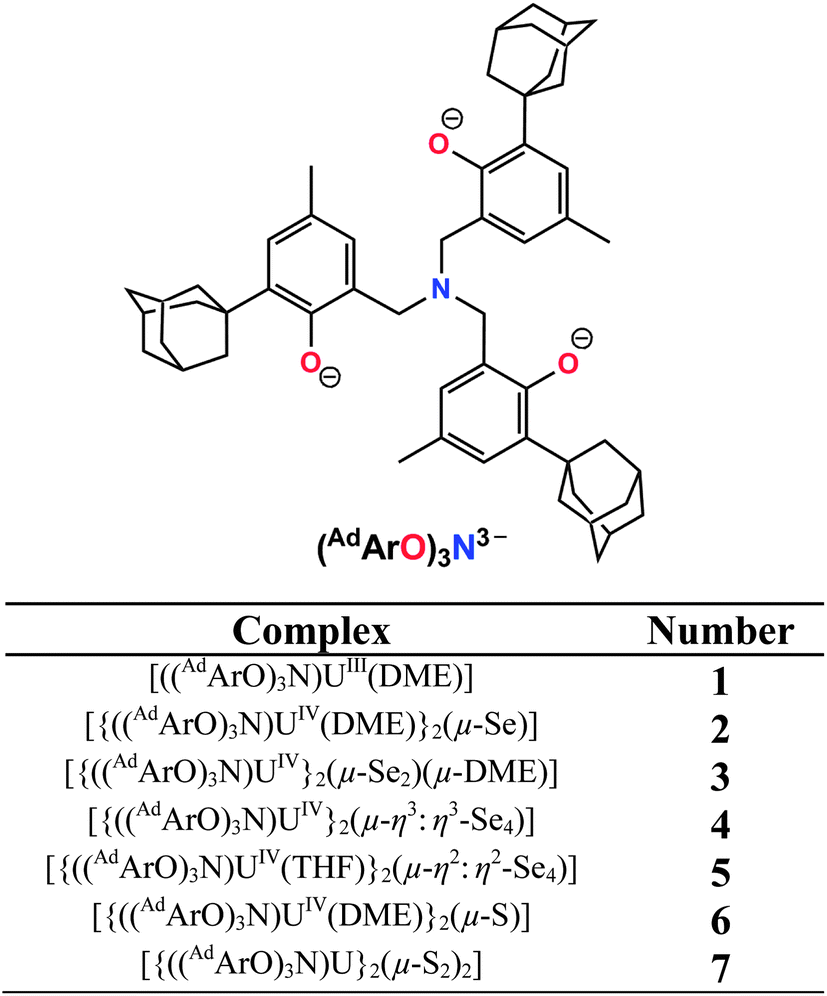
| | Chart 1 Complex formulas and numbering scheme of uranium complexes 1–7, employing the chelating N-anchored ligand (AdArO)3N3− (top). | |
Syntheses and molecular structures of di- and tetraselenido bridged uranium complexes
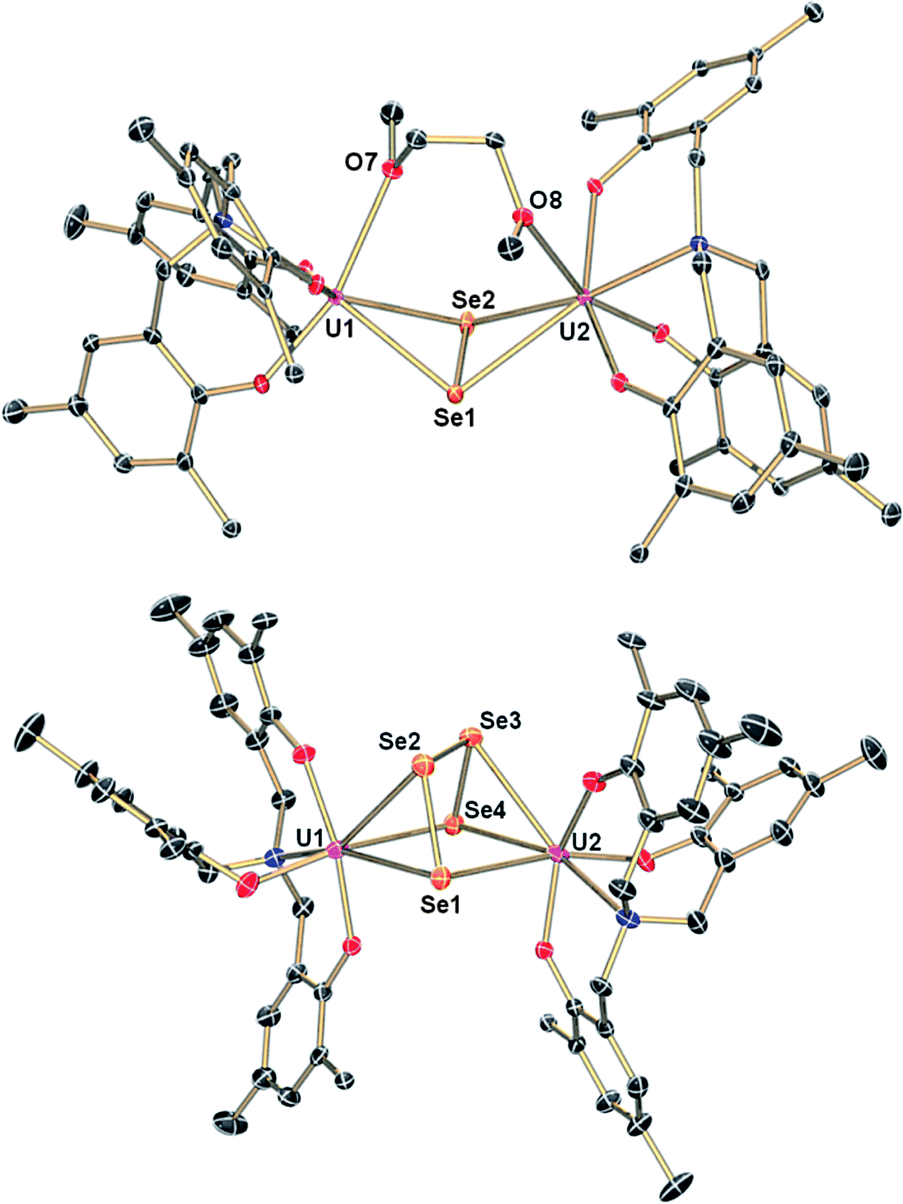
The reaction of a yellow solution of the dinuclear, tetravalent complex [{((AdArO)3N)UIV(DME)}2(μ-Se)] (2) in benzene with precisely 1 equiv. of elemental selenium powder results in the formation of a dark brown solution after several hours. Filtration of the reaction solution and subsequent removal of the volatiles yields the complex [{((AdArO)3N)UIV}2(μ-Se2)(μ-DME)] (3) in 85% yield (Scheme 1, right). This reactivity is remarkable since the uranium(IV) oxidation state, as well as the heavy chalcogens, are considered to be rather unreactive, and the requirements for elemental chalcogen activation usually are either a coordinatively unsaturated, strongly-reducing metal complex or a compound with a metal–metal bond.32 Nonetheless, Hayton et al. were able to perform the remarkable two-electron oxidation of the terminal monooxo uranium(IV) complex [Cp*2Co][(R2N)3U(O)] (R = SiMe3) with the elemental chalcogens to form sulfur and selenium substituted uranyl analogues [OUE]2+ (E = S, Se).22 Also, the choice of an appropriate ligand appears to play a key role, since even iodine can oxidize U(IV) to U(V) under the right conditions.42 The bridging Se2− ligand in the uranium(IV) starting complex [{((AdArO)3N)UIV(DME)}2(μ-Se)] (2) apparently is very electron-rich and thus capable of reducing another selenium atom without involving the two uranium(IV) centers in any further redox chemistry. Alternatively, 3 can be synthesized directly from the uranium(III) starting material [((AdArO)3N)UIII(DME)] and precisely 2 equiv. of selenium powder (Scheme 1). Recrystallization of 3 in a concentrated DME solution yields brown crystals suitable for X-ray diffraction. The solid state structure reveals the dinuclear, tetravalent uranium complex [{((AdArO)3N)UIV}2(μ-Se2)(μ-DME)] that features the bridging μ-η2:η2-Se22− unit as well as a bridging DME molecule (Fig. 1, top). The coordination geometry of both seven-coordinate uranium centers can be best described as distorted mono-capped trigonal prismatic. The η2-bridging Se22− unit is located in the axial position and is trans to the nitrogen anchor. This geometry is in contrast to complex 2, where the bridging μ-Se unit is located in the equatorial position cis to the nitrogen anchor.14 The U–Se bonds are slightly asymmetric (U1–Se1, 2.962(1) Å; U1–Se2, 3.060(1) Å; U2–Se1, 3.078(1) Å; U2–Se2, 2.942(1) Å) and are slightly longer compared to typical U–Se–U bond distances (Table 1).14,19,37,43 The Se1–Se2 bond distance of 2.377(1) Å is comparable to the respective bond lengths in transition metal32,33,44,45 and lanthanide complexes32,46–49 as well as uranium perselenide complexes (2.293(2) Å–2.401(4) Å).39,50 Additionally, the U(IV)–Se22−–U(IV) entity adopts a M2L2-butterfly structural motif, with a torsion angle of 125.02° and is thus strongly bent compared to the structurally similar bis-μ-selenido complex [Na(DME)3]2[{((AdArO)3N)UIV}2(μ-Se)2] that has a torsion angle of 160.8° between the two uranium centers and the two Se2− ligands.14 The average U–OAr bond distances of 2.149 Å are slightly shorter than those observed in 2 (2.178, 2.179 Å) but are still within the range of U–OAr bond distances in other complexes supported by the (AdArO)3N3− ligand.14,20,21
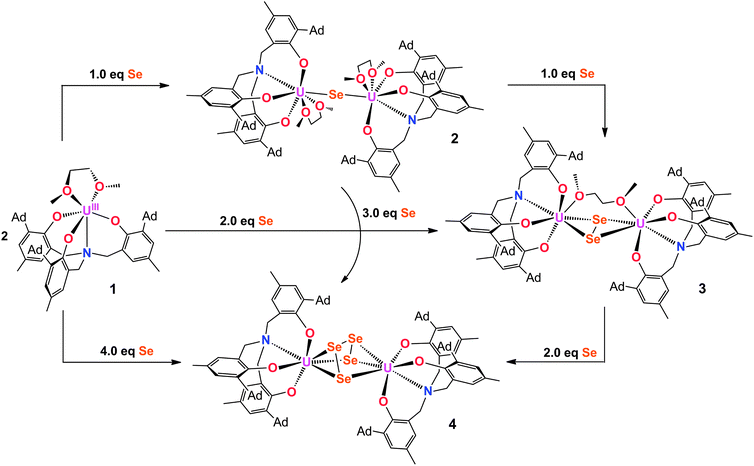 |
| | Scheme 1 Syntheses of selenium-bridged complexes 2 (top), 3 (right), and 4 (bottom) via different pathways using precise stoichiometric amounts of elemental selenium. | |
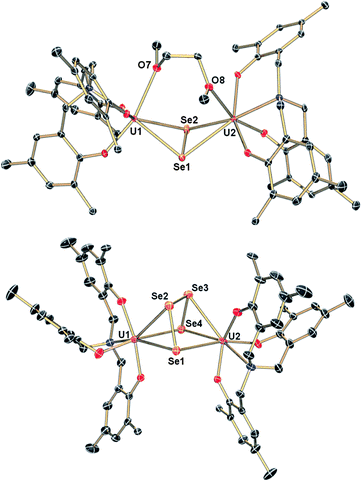 |
| | Fig. 1 Molecular structures of uranium diselenido complex 3 (top) in crystals of 3·2DME and uranium tetraselenido complex 4 (bottom) in crystals of 4·2C7H8·0.25C6H14. Adamantyl groups, H-atoms and co-crystallized solvent molecules are omitted for clarity. Thermal ellipsoids are at 50% probability. | |
Table 1 Selected bond distances for complexes 2–5 (in Å)
| Structural parameter |
2 (ref. 14) |
3
|
4
|
5
|
| U1–Se |
2.830(1) |
2.962(1), 3.060(1) |
3.076(1), 3.121(1), 3.052(2) |
2.913(1), 3.178(1) |
| U2–Se |
2.816(1) |
3.079(1), 2.942(1) |
3.056(1), 3.125(1), 3.077(2) |
3.178(1), 2.913(1) |
| Se1–Se2, Se3–Se4 |
— |
2.377(1) |
2.316(2), 2.310(2) |
2.288(1) |
| Se2–Se3 |
— |
— |
2.438(1) |
2.420(2) |
| U–OAr |
2.178, 2.179 |
2.149, 2.164 |
2.123, 2.126 |
2.135 |
| U1,2–N1,2 |
2.556(6), 2.551(6) |
2.595(2), 2.601(2) |
2.545(3), 2.551(3) |
2.591(4) |
In attempts to further investigate the synthesis of complex 3, we added increasingly more than 2 equiv. of Se, ultimately leading to the formation of yet another, olive-green product. An X-ray diffraction analysis of single crystals obtained by slow diffusion of hexane into a saturated toluene solution revealed a dinuclear tetravalent uranium complex, namely [{((AdArO)3N)UIV}2(μ-η3:η3-Se4)] (4); now bridged by four selenium atoms with a formal oxidation state of Se42− (Fig. 1, bottom). Hence, we adjusted the reaction conditions appropriately and added exactly 4 equiv. of selenium powder to [((AdArO)3N)UIII(DME)] and stirred the reaction solution for 3 days after which olive-green 4 forms (Scheme 1, bottom). Complex 4 was obtained after filtration and drying of the solids in vacuo in 90% yield. Alternatively, [{((AdArO)3N)UIV(DME)}2(μ-Se)] (2) can be treated with just 3 equiv. of selenium powder to obtain 4 with a similar yield. Evidently, 3 is still reactive towards elemental selenium powder and is able to insert two more selenium atoms into the bridging Se22− unit of 3 to form the Se42− ligand found in 4. This reactivity clearly shows the proclivity of selenium to catenate to chains as there is no redox chemistry involving the uranium centers and their oxidation state of +IV is retained (vide infra). The molecular structure of 4 shows two tetravalent uranium centers in a distorted mono-capped trigonal prismatic geometry. Two Se atoms of the Se42− moiety are located trans and one Se atom is situated cis to the nitrogen anchor. In contrast to 3, the uranium centers in 4 are not coordinated by additional solvent molecules. The bridging Se42− moiety features alternating Se–Se bond distances with two very similar bond lengths with Se1–Se2 at 2.316(2) and Se3–Se4 at 2.310(2) Å as well as a slightly larger bond distance of 2.438(1) Å for Se2–Se3 (Table 1). Thus, the bridging Se42− unit is reminiscent to complexes of the 1,3-butadiene ligand.51–53 Similar bond properties are known for polysulfide ligands and can be attributed to charge localization at the first and last selenium atom of the chain.33,54,55 Only three selenium atoms of the Se42− ligand are coordinated to each uranium center in a μ-η3:η3 fashion. The U–Se bonds are all slightly asymmetric and range from 3.052(2) to 3.125(1) Å, which is marginally longer than the bond distances in 3 and other reported U–Se distances.8,14,19 The average U–OAr bond distances of 2.123 and 2.126 Å in 4 are even shorter than those observed in 3 (2.149, 2.164 Å), which indicates a stronger aryloxide-to-uranium binding and lower electron density at the uranium centers with an increasing number of coordinated Se atoms. It should be noted that the crystal structure of the investigated crystal of 4 revealed the presence of a minor fraction (less than 7%) of the corresponding bis-μ-selenido complex [{((AdArO)3N)U}2(μ-Se)2] leading to some disorder in the central U–Se4–U moiety.56
Crystallization of 4 from coordinating solvent induces a geometry change of the coordinated Se42− ligand. X-ray diffraction analysis of olive-green/brown single crystals, obtained from slow hexane diffusion into a THF solution of the complex reveal the molecular structure of 5 (Fig. 2, Scheme 2). The dinuclear tetravalent uranium complex [{((AdArO)3N)UIV(THF)}2(μ-η2:η2-Se4)] features a bridging Se42− ligand chain (as opposed to the open-square-like Se42− unit in 4). Additionally, both uranium centers are now coordinated by one additional THF solvent molecule. In contrast to 4, the bridging Se42− unit of 5 is coordinated in a μ-η2:η2-fashion and is structurally very similar to the bridging tetraselenido complex [(tBu2bpy)U(NtBu)2(I)]2(μ-η2:η2-Se4) synthesized by Boncella et al.37 The dinuclear complex is situated on a crystallographic inversion center and exhibits Ci molecular symmetry. Each uranium center in 5 adopts a distorted mono-capped trigonal prismatic coordination geometry. The U–Se bond lengths of 2.913(1) Å and 3.178(1) Å are indicative of a strong anionic U–Se1 bond and a weaker dative interaction U–Se2, respectively, an observation that is identical to that reported by Boncella and coworkers.37 Furthermore, similar to 4, compound 5 exhibits two shorter Se–Se bonds at 2.288(1) Å and one longer bond at 2.420(0) Å between Se2–Se3; the average U–OAr bond distances of 2.135 Å are comparable with those observed for compound 4 (Table 1). Complexes 4 and 5 are unambiguously distinguishable by 1H NMR spectroscopy. The signals of 4 are very sharp compared to the broadened signals of 5, which is likely due to the fluxional nature of the coordinated THF solvent molecules. It should be noted that a second minor species (5.5%) in the investigated crystal of 5 was identified as [{((AdArO)3N)UIV(THF)}2(μ-η2:η2-Se3)] with a bridging Se32− ligand (see ESI†).56 Unfortunately, this compound could not be synthesized reproducibly nor isolated in pure form. Attempts to isolate different products by adding even more equivalents of elemental selenium to compounds 4 and 5 have not been successful. This observation might be attributed to the decreasing solubility of complexes 4 and 5.
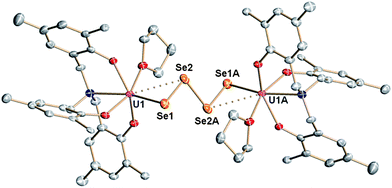 |
| | Fig. 2 Molecular structure of THF-coordinated uranium tetraselenido complex 5 in crystals of 5·2C7H8. Adamantyl groups, H-atoms and co-crystallized solvent molecules are omitted for clarity. Thermal ellipsoids are at 50% probability. | |
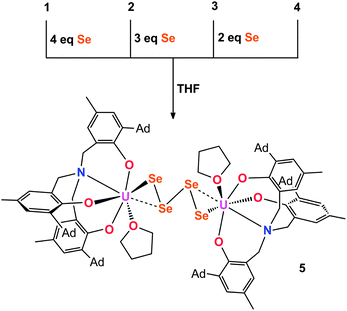 |
| | Scheme 2 Synthesis of THF-coordinated tetraselenido complex 5 from different starting materials 1–4. | |
Synthesis and molecular structure of polysulfido-bridged uranium complexes
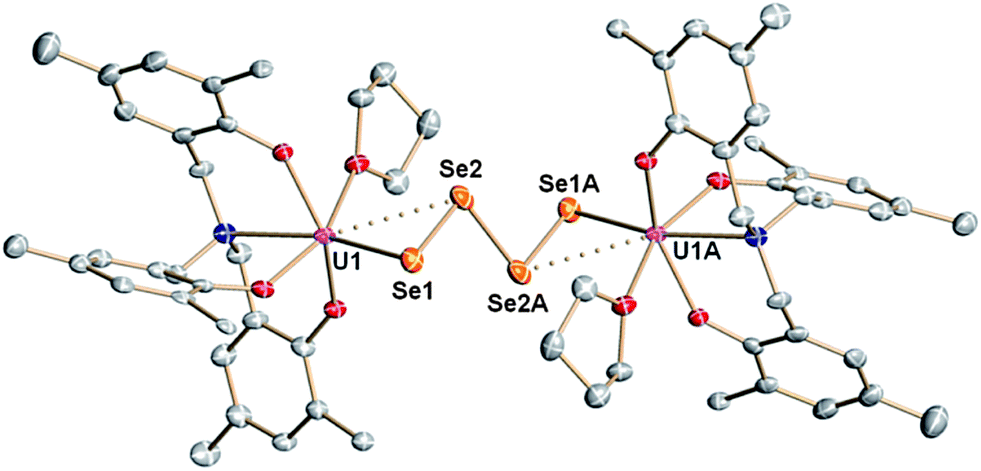
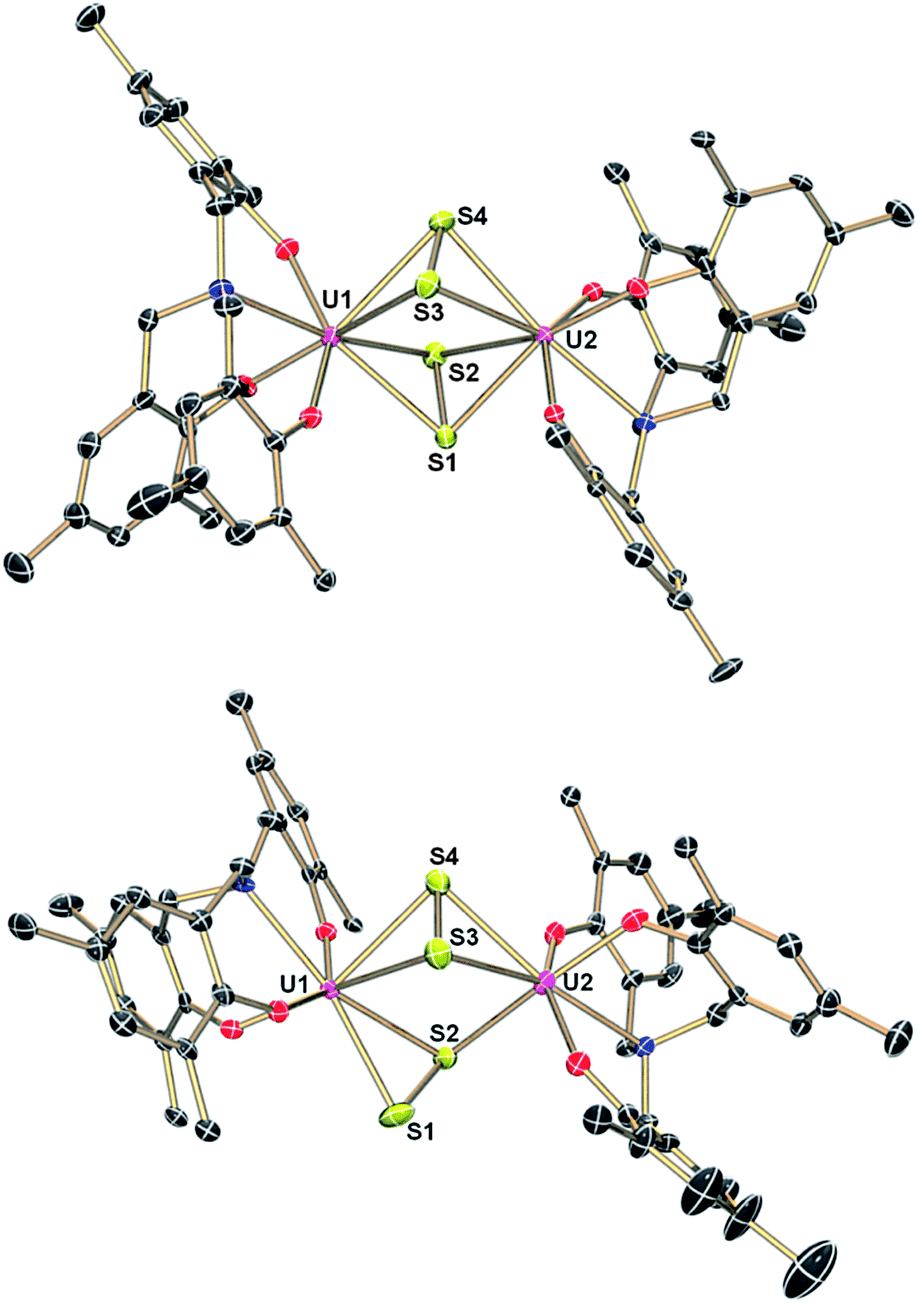
The μ-sulfido complex [{((AdArO)3N)UIV(DME)}2(μ-S)] (6) exhibits similar reactivity to the μ-selenido bridged complex [{((AdArO)3N)UIV(DME)}2(μ-Se)] (2) if treated with 0.375 equiv. of elemental sulfur in benzene (Scheme 3). Immediate colour change of the reaction mixture to dark brown was observed and, within 5 minutes of further stirring, a dark brown solid precipitated. After 2 hours of stirring, the solution was filtered and the precipitate was washed with benzene and dried in vacuo to yield complex 7 analytically pure in 88% yield. An X-ray diffraction analysis of crystals grown by diffusing DME into a saturated THF solution revealed two crystallographically independent but chemically equivalent molecules in the asymmetric unit (Z = 2), namely two dinuclear uranium complexes [{((AdArO)3N)U}2(μ-η2:η2-S2)2] (7A) (Fig. 3, top) and [{((AdArO)3N)U}2(μ-η2:η2-S2)(μ-η1:η2-S2)] (7B) (Fig. 3, bottom) that contain four bridging sulfur atoms. However, in contrast to 4, the bridging unit in both complexes 7A and 7B is not comprised of a S42− chain-type ligand but of two independent S2 moieties. In contrast to complexes 3–5, and within the scope of the present study, the oxidation state of the uranium centers (and chalcogenide ligands) in 7A and 7B cannot be unambiguously identified (vide infra). Complexes 7A and 7B could be formulated either as two U(IV) centers with two bridging supersulfido S2− units or as two U(V) centers bridged by two persulfido S22− ligands. Each of the eight-coordinate uranium centers in 7A adopts a distorted square-antiprismatic coordination geometry. The bridging S2 fragments in 7A both feature an η2:η2-binding mode and show two almost identical S1–S2 and S3–S4 bond distances of 2.050(2) Å and 2.053(2) Å, respectively (Table 2). These bond distances are at the shorter end of known S–S bond lengths observed in other uranium1,19,50,57–61 and transition metal complexes,62–66 featuring the persulfido ligand S22−. However, the very rare supersulfido complexes of the transition metals have shown a wider range for the S–S bond length, ranging from 1.944–2.023 Å, depending on the metal center, the supporting ligand, and the coordination mode.67–70 Accordingly, based on the bond metric of the S2 moiety, an exact assignment (persulfide vs. supersulfide) remains impossible. Both ligands are slightly skewed, pointing toward each other, and the distances between S1–S3 (3.836 Å) and S2–S4 (3.366 Å) rule out the formation of a conceivable S42− unit. The U–S bond distances are slightly different from each other and range from 2.741(2) to 3.020(2) Å, which is slightly longer than the bond lengths observed in [{((AdArO)3N)UIV(DME)}2(μ-S)]14 (6) and other bridging sulfido complexes reported in the literature.6,19,37 The average U–OAr bond distances of 2.104 Å and 2.106 Å, respectively, are significantly shorter than those observed in 6, which is an observation that was also made for complexes 4 and 5. Complex 7B is slightly different from 7A as only one S2 unit features the η2:η2-binding mode that was already observed in 7A. The second S2 moiety exhibits a μ-η1:η2 binding mode, which results in both uranium centers being situated in a different coordination environment. The U1 ion has a coordination number of eight and features a distorted square-antiprismatic coordination geometry, while U2 is only seven-coordinate and adopts a distorted mono-capped octahedral coordination environment. The U–S bond lengths (2.684(2)–2.860(2) Å) and S–S bond distances (2.051(2), 2.044(2) Å) are all in the same range as observed in 7A (Table 2). Finally, the average U–OAr bond distances of 2.110 Å and 2.087 Å can be compared to the observations made in complexes 4, 5, and 7A. We investigated if the persulfido complex [{((AdArO)3N)UIV}2(μ-S2)], which might be a possible intermediate in the formation of 7, could be synthesized similarly to 3. However, all attempts to form a persulfido complex lead to the precipitation of 7. This selective formation of 7 likely is due to the S8 ring structure of elemental sulfur as well as the low solubility of complex 7, which leads to an excess of sulfur atoms in the coordination vicinity of the uranium center and subsequent precipitation of 7. Likewise, trying to form the persulfido complex by reacting 7 with stoichiometric amounts of 1 in a comproportionation reaction only lead to intractable mixtures of different products, from which no pure compound could be isolated.
 |
| | Scheme 3 Syntheses of sulfur-bridged complexes 6 (top) and 7 (bottom) via different pathways using precise stoichiometric amounts of elemental sulfur. A disulfido complex (right) is proposed as an intermediate during the formation of 7. | |
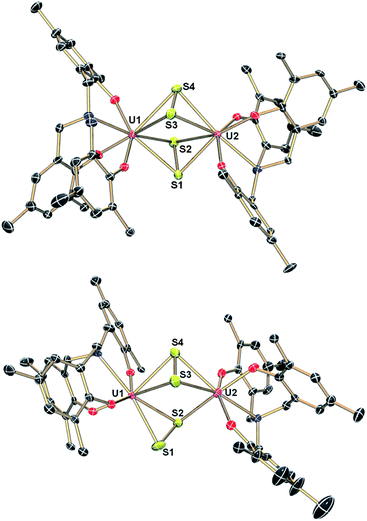 |
| | Fig. 3 Molecular structures of uranium disulfide complex 7A (top) and uranium disulfide complex 7B (bottom) in crystals of 7·5DME. Adamantyl groups, H-atoms and co-crystallized solvent molecules are omitted for clarity. Thermal ellipsoids are at 50% probability. | |
Table 2 Selected bond distances (in Å) for complexes 6, 7A, and 7B
| Structural parameter |
6 (ref. 14) |
7A
|
7B
|
| U1–S |
2.736(2) |
2.741(2), 3.020(2), 2.809(2), 2.767(2) |
2.684(2), 2.860(2), 2.842(2), 2.859(2) |
| U2–S |
2.713(2) |
2.858(2), 2.763(2), 2.766(2), 2.924(2) |
2.724(2), 2.783(2), 2.757(2) |
| S1–S2, S3–S4 |
— |
2.050(2), 2.053(2) |
2.051(2), 2.044(2) |
| U–OAr |
2.185, 2.180 |
2.104, 2.106 |
2.110, 2.087 |
| U1,2–N1,2 |
2.558(6), 2.565(6) |
2.550(4), 2.529(4) |
2.566(4), 2.554(4) |
Electronic structure of polychalcogenido bridged U(IV/IV) complexes
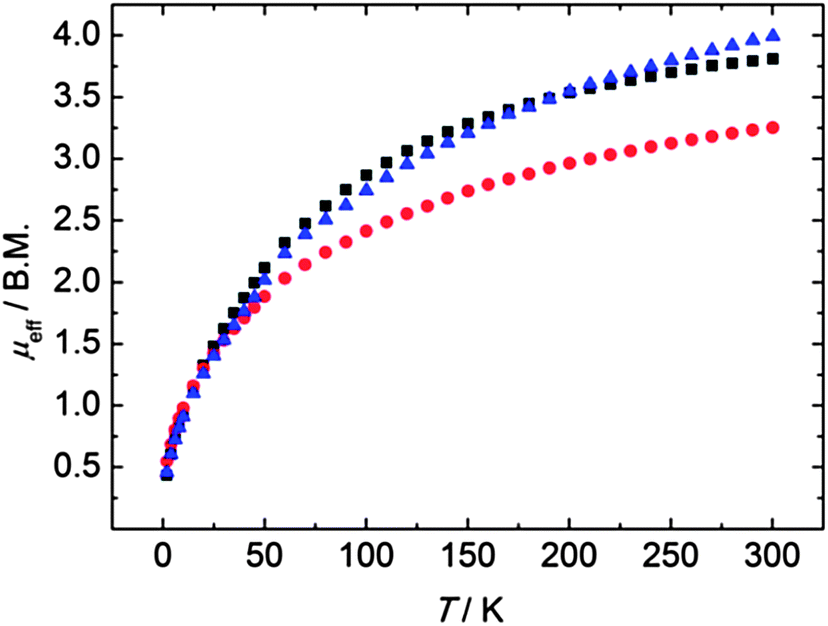
In order to probe the formal oxidation state of the uranium ions and their electronic structures, we performed variable temperature (2–300 K) dc magnetization measurements on compounds 3, 4, and 5. SQUID measurements reveal magnetic moments of μeff = 3.81 (3), 3.25 (4) and 3.99 (5) μB at 300 K, which decrease with decreasing temperature to μeff = 0.43 (3), 0.55 (4), and 0.46 (5) μB at 2 K (Fig. 4). Accordingly, the observed magnetic moments and their temperature-dependency is characteristic of uranium(IV) complexes with an f2 electron configuration and a 3H4 ground state.14,21,71
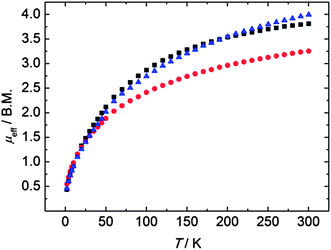 |
| | Fig. 4 Temperature-dependent SQUID magnetization data of polyselenido-bridged compounds 3 (black), 4 (red), and 5 (blue). Data were corrected for underlying diamagnetism. | |
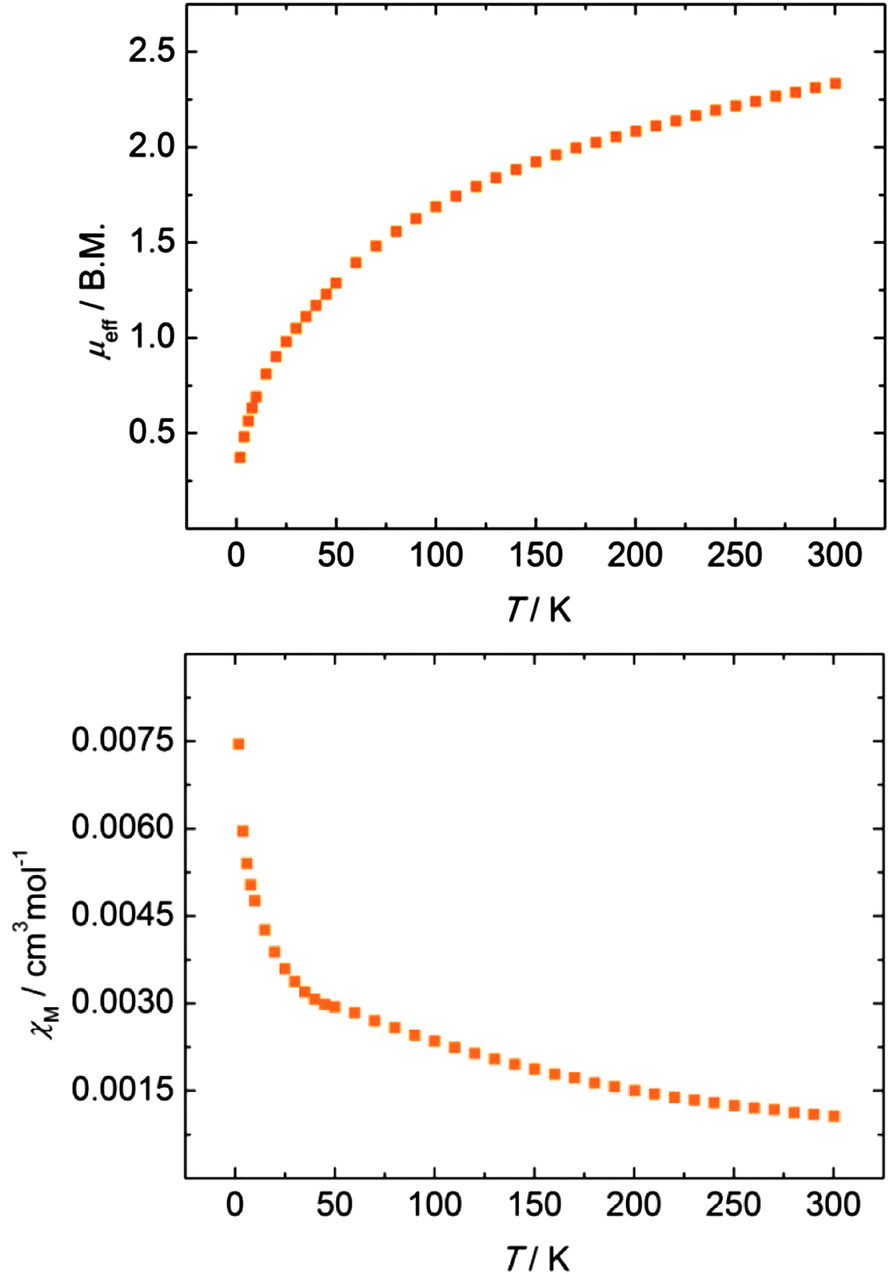
An investigation of the temperature-dependent behaviour of disulfido bridged complex 7 revealed a magnetic moment of μeff = 2.33 μB at 300 K (per formula unit) that decreases to 0.37 μB at 2 K upon decreasing temperature (Fig. 5, top). While a low-temperature magnetic moment of ∼0.4–0.5 μB is typically observed in uranium(IV) compounds, the unusually low magnetic moment of 2.33 μB at room temperature is more indicative of a uranium(V) species.72 Antiferromagnetic exchange coupling between two uranium centers can significantly decrease the overall magnetic moment and is typically observed as a maximum in the plot of χMvs. T.14,73 The magnetic susceptibility of 7, however, does not show a maximum (Fig. 5, bottom); and hence, antiferromagnetic exchange coupling is not evident for this compound but cannot be ruled out entirely. Furthermore, increased covalency of the U–Se bonds in 3–5 and the U–S bonds in 7, respectively, can also reduce the overall magnetic moment.74 The observed non-magnetic ground state of 7 at low temperature (μeff = 0.37 B.M. at 2 K) allows for different interpretations, arising from several possible electronic configurations and magnetic exchange interactions within the [U(S2)2U] unit: the ground state could result from non-interacting U(IV) f2 ions (singlet Γ1 ground state within the 3H4 ground manifold) with through-space, antiferromagnetically coupled supersulfide (S2˙−) ligands or an entirely antiferromagnetically coupled system of U(IV) and bridging radical anions. Alternatively, and assuming that the bridging S2 ligands are further reduced to diamagnetic persulfides, S22−, the U(V) f1 ions could be strongly antiferromagnetically coupled to yield the observed non-magnetic ground state at 2 K. Closer inspection of the χMvs. T plot (Fig. 5, bottom) indeed reveals a plateau at approx. 50 K with increasing χM below 30 K. While the increased susceptibility at lower temperatures might arise from minor paramagnetic impurities, the saddle point could – in principle – be qualitatively interpreted as the onset of an ordering phenomenon in an antiferromagnetically coupled system.73,75–77 However, the current magnetization study does not allow for an unambiguous assignment of the U ions' and bridging S2 ligands' oxidation states.
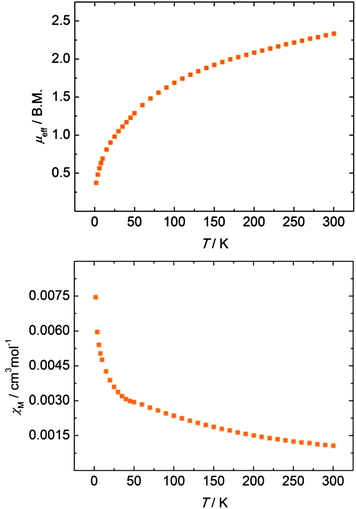 |
| | Fig. 5 Temperature-dependent SQUID magnetization data of polysulfido-bridged compound 7 as a plot of μeffvs. T (top) and χMvs. T (bottom). Data were corrected for underlying diamagnetism. | |
Accordingly, CW X-band EPR spectroscopic studies were performed on samples of 7 in liquid and frozen toluene solution at room temperature and 7 K, respectively. However, samples of 7 are EPR silent under these conditions. This is not unusual and – in principle – not in contrast with the formulation of 7 as U(IV/IV) with two bridging, radical anionic, supersulfido ligands, nor as paramagnetic U(V/V) with bridging, diamagnetic persulfido ligands. Previous studies have shown that both U(V) species (e.g., U(V) imidos)78 and complexes of U(IV) with coordinated radical anions, such as carbon dioxide,79 diphenyldiazomethane,80 or benzophenone81 are EPR inactive. These observations render attempts to assign the exact oxidation state of the uranium ions as well as the bridging ligands in complexes 7A and 7B difficult. Full understanding of the magnetic behavior of this compound and comparison to 1–6 requires collaborative XAS spectroscopic and DFT theoretical studies that are currently being planned and will be reported in due time.
Conclusion
Polychalcogenido complexes of actinides – especially those featuring the heavier group 16 elements S, Se, and Te – are an exceedingly rare class of compounds. Generally, low-valent, strongly-reducing metals are essential for the activation of elemental chalcogens. Herein we demonstrated, however, that by carefully choosing the reaction conditions, even relatively unreactive uranium(IV) complexes can be employed for controlled and stoichiometric elemental chalcogen activation; given the chelating ligand at the U(IV) ion provides sufficient stability and flexibility. Thus, employing the sterically highly accommodating (AdArO)3N3− chelate, polyselenido complexes 3, 4, and 5 were synthesized via different pathways, starting from either the U(IV) complex (2 and 6) or the U(III) complex (1), respectively. These complexes feature μ-Se22−, μ-η3:η3-Se42−, and μ-η2:η2-Se42− bridging ligands. Selectivity of compound formation is achieved by using precise stoichiometric amounts of elemental chalcogen and appropriate solvents. The resulting complexes showed magnetic behavior that is characteristic of the uranium(IV) starting compounds; thus, demonstrating the unchanged redox state of the uranium centers. Hence, the reactivity likely stems from the Se2− and Se22− moieties themselves and is independent from the metal centers that are basically serving as a stage for chalcogenide transformation. The reactivity of elemental sulfur with the respective U(III) and U(IV) complexes is distinctly different from that of selenium. The resulting complex [{((AdArO)3N)U}2(μ-S2)2] (7) features two bridging S2 units instead of a single S42− ligand as observed with selenium. These novel uranium polychalcogenido complexes are currently being investigated with respect to chalcogen atom transfer chemistry.
Acknowledgements
This work was supported by funds from the German Federal Ministry of Education and Research (support codes 02 NUK 012C and 02 NUK 020C). We thank Dr. Andreas Scheurer for performing the VT NMR experiments.
Notes and references
- D. L. Perry, A. Zalkin, H. Ruben and D. H. Templeton, Inorg. Chem., 1982, 21, 237–240 CrossRef CAS.
- D. A. Wrobleski, D. T. Cromer, J. V. Ortiz, T. B. Rauchfuss, R. R. Ryan and A. P. Sattelberger, J. Am. Chem. Soc., 1986, 108, 174–175 CrossRef CAS.
- C. Lescop, T. Arliguie, M. Lance, M. Nierlich and M. Ephritikhine, J. Organomet. Chem., 1999, 580, 137–144 CrossRef CAS.
- L. Ventelon, C. Lescop, T. Arliguie, M. Ephritikhine, P. C. Leverd, M. Lance and M. Nierlich, Chem. Commun., 1999, 659–660 RSC.
- B. Liang, L. Andrews, N. Ismail and C. J. Marsden, Inorg. Chem., 2002, 41, 2811–2813 CrossRef CAS PubMed.
- J. G. Brennan, R. A. Andersen and A. Zalkin, Inorg. Chem., 1986, 25, 1761–1765 CrossRef CAS.
- L. R. Avens, D. M. Barnhart, C. J. Burns, S. D. McKee and W. H. Smith, Inorg. Chem., 1994, 33, 4245–4254 CrossRef CAS.
- A. J. Gaunt, B. L. Scott and M. P. Neu, Inorg. Chem., 2006, 45, 7401–7407 CrossRef CAS PubMed.
- W. J. Evans, E. Montalvo, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Inorg. Chem., 2009, 49, 222–228 CrossRef PubMed.
- W. J. Evans, K. A. Miller, S. A. Kozimor, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2007, 26, 3568–3576 CrossRef CAS.
- W. J. Evans, M. K. Takase, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2008, 28, 236–243 CrossRef.
- C. R. Graves, B. L. Scott, D. E. Morris and J. L. Kiplinger, Chem. Commun., 2009, 776–778 RSC.
- X. Wang, L. Andrews and C. J. Marsden, Inorg. Chem., 2009, 48, 6888–6895 CrossRef CAS PubMed.
- O. P. Lam, F. W. Heinemann and K. Meyer, Chem. Sci., 2011, 2, 1538–1547 RSC.
- O. P. Lam, F. W. Heinemann and K. Meyer, Angew. Chem., Int. Ed., 2011, 50, 5965–5968 CrossRef CAS PubMed.
- W. Ren, G. Zi, D.-C. Fang and M. D. Walter, J. Am. Chem. Soc., 2011, 133, 13183–13196 CrossRef CAS PubMed.
- N. A. Siladke, J. W. Ziller and W. J. Evans, J. Am. Chem. Soc., 2011, 133, 3507–3516 CrossRef CAS PubMed.
- R. Thomson, C. Graves, B. Scott and J. Kiplinger, J. Chem. Crystallogr., 2011, 41, 1241–1244 CrossRef CAS PubMed.
- J. L. Brown, G. Wu and T. W. Hayton, Organometallics, 2012, 32, 1193–1198 CrossRef.
- O. P. Lam, L. Castro, B. Kosog, F. W. Heinemann, L. Maron and K. Meyer, Inorg. Chem., 2012, 51, 781–783 CrossRef CAS PubMed.
- O. P. Lam, S. M. Franke, F. W. Heinemann and K. Meyer, J. Am. Chem. Soc., 2012, 134, 16877–16881 CrossRef CAS PubMed.
- J. L. Brown, S. Fortier, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2013, 135, 5352–5355 CrossRef CAS PubMed.
- A. Stein, S. W. Keller and T. E. Mallouk, Science, 1993, 259, 1558–1564 CAS.
- R. H. Bube, Annu. Rev. Mater. Sci., 1990, 20, 19–50 CrossRef CAS.
- S. Lange and T. Nilges, Chem. Mater., 2006, 18, 2538–2544 CrossRef CAS.
- N. Zheng, X. Bu and P. Feng, Nature, 2003, 426, 428–432 CrossRef CAS PubMed.
- J. R. Sootsman, H. Kong, C. Uher, J. J. D'Angelo, C.-I. Wu, T. P. Hogan, T. Caillat and M. G. Kanatzidis, Angew. Chem., Int. Ed., 2008, 47, 8618–8622 CrossRef CAS PubMed.
- H. Xu, K. M. Kleinke, T. Holgate, H. Zhang, Z. Su, T. M. Tritt and H. Kleinke, J. Appl. Phys., 2009, 105, 053703–053705 CrossRef PubMed.
- N. D. Lowhorn, T. M. Tritt, E. E. Abbott and J. W. Kolis, Appl. Phys. Lett., 2006, 88, 022101–022103 CrossRef PubMed.
- L. C. Roof and J. W. Kolis, Chem. Rev., 1993, 93, 1037–1080 CrossRef CAS.
- C. Graf, A. Assoud, O. Mayasree and H. Kleinke, Molecules, 2009, 14, 3115–3131 CrossRef CAS PubMed.
- M. G. Kanatzidis and S.-P. Huang, Coord. Chem. Rev., 1994, 130, 509–621 CrossRef CAS.
- W. S. Sheldrick, Z. Anorg. Allg. Chem., 2012, 638, 2401–2424 CrossRef CAS.
- C. Madic, M. Lecomte, P. Baron and B. Boullis, C. R. Phys., 2002, 3, 797–811 CrossRef CAS.
- T. Mehdoui, J.-C. Berthet, P. Thuery and M. Ephritikhine, Chem. Commun., 2005, 2860–2862 RSC.
- K. I. M. Ingram, N. Kaltsoyannis, A. J. Gaunt and M. P. Neu, J. Alloys Compd., 2007, 444–445, 369–375 CrossRef CAS PubMed.
- L. P. Spencer, P. Yang, B. L. Scott, E. R. Batista and J. M. Boncella, Inorg. Chem., 2009, 48, 11615–11623 CrossRef CAS PubMed.
- J. L. Brown, S. Fortier, R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 15468–15475 CrossRef CAS PubMed.
- A. C. Sutorik and M. G. Kanatzidis, J. Am. Chem. Soc., 1991, 113, 7754–7755 CrossRef CAS.
- O. P. Lam, S. C. Bart, H. Kameo, F. W. Heinemann and K. Meyer, Chem. Commun., 2010, 46, 3137–3139 RSC.
- J. E. McDonough, A. Mendiratta, J. J. Curley, G. C. Fortman, S. Fantasia, C. C. Cummins, E. V. Rybak-Akimova, S. P. Nolan and C. D. Hoff, Inorg. Chem., 2008, 47, 2133–2141 CrossRef CAS PubMed.
- O. J. Cooper, D. P. Mills, J. McMaster, F. Moro, E. S. Davies, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 2383–2386 CrossRef CAS PubMed.
- W. J. Evans, E. Montalvo, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Inorg. Chem., 2010, 49, 222–228 CrossRef CAS PubMed.
- M. Shieh, C.-H. Ho, W.-S. Sheu and H.-W. Chen, J. Am. Chem. Soc., 2010, 132, 4032–4033 CrossRef CAS PubMed.
- J. Matsumoto, Y. Kajita and H. Masuda, Inorg. Chem., 2012, 51, 1236–1238 CrossRef CAS PubMed.
- L. Huebner, A. Kornienko, T. J. Emge and J. G. Brennan, Inorg. Chem., 2005, 44, 5118–5122 CrossRef CAS PubMed.
- A. Kornienko, J. H. Melman, G. Hall, T. J. Emge and J. G. Brennan, Inorg. Chem., 2002, 41, 121–126 CrossRef CAS PubMed.
- J. Liang, J. Chen, J. Zhao, Y. Pan, Y. Zhang and D. Jia, Dalton Trans., 2011, 40, 2631–2637 RSC.
- W. J. Evans, G. W. Nyce, R. D. Clark, R. J. Doedens and J. W. Ziller, Angew. Chem., Int. Ed., 1999, 38, 1801–1803 CrossRef CAS.
- E. M. Matson, M. D. Goshert, J. J. Kiernicki, B. S. Newell, P. E. Fanwick, M. P. Shores, J. R. Walensky and S. C. Bart, Chem.–Eur. J., 2013, 19, 16176–16180 CrossRef CAS PubMed.
- M. R. Churchill and J. Wormald, Inorg. Chem., 1969, 8, 1936–1941 CrossRef CAS.
- H. P. Xia, R. C. Y. Yeung and G. Jia, Organometallics, 1998, 17, 4762–4768 CrossRef CAS.
- M.-C. Chung, X. Gu, B. A. Etzenhouser, A. M. Spuches, P. T. Rye, S. K. Seetharaman, D. J. Rose, J. Zubieta and M. B. Sponsler, Organometallics, 2003, 22, 3485–3494 CrossRef CAS.
- C. Müller and P. Böttcher, Z. Naturforsch. B, 1993, 48, 1732 Search PubMed.
- C. Müller and P. Böttcher, Z. Naturforsch. B, 1994, 49, 489 Search PubMed.
- The impurities observed and refined in multiple single crystal X-ray diffraction studies of complexes 4 and 5 cannot be identified in the 1H NMR spectra of freshly prepared samples of these compounds; hence, concomitant formation can be ruled out. It is likely that the impurities are formed when complexes 4 and 5 are in solution for a prolonged period of time, such as during crystallization.
- A. C. Sutorik and M. G. Kanatzidis, Polyhedron, 1997, 16, 3921–3927 CrossRef CAS.
- D. J. Grant, Z. Weng, L. J. Jouffret, P. C. Burns and L. Gagliardi, Inorg. Chem., 2012, 51, 7801–7809 CrossRef CAS PubMed.
- J.-E. Kwak, D. L. Gray, H. Yun and J. A. Ibers, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, i86–i87 CAS.
- D. E. Bugaris and J. A. Ibers, Dalton Trans., 2010, 39, 5949–5964 RSC.
- M. Mazzanti, J. Pecaut, C. Camp, I. Ciofini, G. Garcia, M. Almeida, M. A. Antunes, J. Marcalo and I. C. Santos, Chem. Sci., 2013 10.1039/C3SC52742E.
- A. Müller, W. Jaegermann and J. H. Enemark, Coord. Chem. Rev., 1982, 46, 245–280 CrossRef.
- S. Ryu, D. Whang, H.-J. Kim, K. Kim, M. Yoshida, K. Hashimoto and K. Tatsumi, Inorg. Chem., 1997, 36, 4607–4609 CrossRef CAS.
- M. N. Sokolov, A. L. Gushchin, S. V. Tkachev, D. Y. Naumov, P. Nuñez, P. Gili, J. G. Platas and V. P. Fedin, Inorg. Chim. Acta, 2005, 358, 2371–2383 CrossRef CAS PubMed.
- M. Rakowski DuBois, B. R. Jagirdar, S. Dietz and B. C. Noll, Organometallics, 1997, 16, 294–296 CrossRef.
- H. Brunner, A. Merz, J. Pfauntsch, O. Serhadli, J. Wachter and M. L. Ziegler, Inorg. Chem., 1988, 27, 2055–2058 CrossRef CAS.
- R. C. Elder and M. Trkula, Inorg. Chem., 1977, 16, 1048–1051 CrossRef CAS.
- A. Terzis and R. Rivest, Inorg. Chem., 1973, 12, 2132–2136 CrossRef CAS.
- J. T. York, E. C. Brown and W. B. Tolman, Angew. Chem., Int. Ed., 2005, 44, 7745–7748 CrossRef CAS PubMed.
- S. Yao, C. Milsmann, E. Bill, K. Wieghardt and M. Driess, J. Am. Chem. Soc., 2008, 130, 13536–13537 CrossRef CAS PubMed.
-
T. H. Siddal, in Theory and Applications of Molecular Paramagnetism, ed. E. A. Bundreaux and L. N. Mulay, John Wiley & Sons, New York, 1976 Search PubMed.
- With the exception of one absorption band observed at approximately 1500 nm (6670 cm−1, ε = 10–100 M−1 cm−1), the UV/vis-NIR spectra of all chalcogenide complexes reported here exhibit broad, featureless absorptions between 400 and 1400 nm (see ESI†). Thus, an absorption feature between 5000 and 6000 cm−1, often indicative of the pentavalent state in uranium complexes,42 is present in all complexes reported here; even for those in which the U(IV) oxidation state is unambiguous.
- R. K. Rosen, R. A. Andersen and N. M. Edelstein, J. Am. Chem. Soc., 1990, 112, 4588–4590 CrossRef CAS.
- C. R. Graves, P. Yang, S. A. Kozimor, A. E. Vaughn, D. L. Clark, S. D. Conradson, E. J. Schelter, B. L. Scott, J. D. Thompson, P. J. Hay, D. E. Morris and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 5272–5285 CrossRef CAS PubMed.
- G. g. Nocton, P. Horeglad, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2008, 130, 16633–16645 CrossRef CAS PubMed.
- V. Mougel, P. Horeglad, G. Nocton, J. Pécaut and M. Mazzanti, Angew. Chem., Int. Ed., 2009, 48, 8477–8480 CrossRef CAS PubMed.
- P. L. Arnold, G. M. Jones, S. O. Odoh, G. Schreckenbach, N. Magnani and J. B. Love, Nat. Chem., 2012, 4, 221–227 CrossRef CAS PubMed.
- I. Castro-Rodriguez, K. Olsen, P. Gantzel and K. Meyer, J. Am. Chem. Soc., 2003, 125, 4565–4571 CrossRef CAS PubMed.
- H. N. I. Castro-Rodriguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. Meyer, Science, 2004, 305, 1757 CrossRef PubMed.
- O. P. Lam, P. L. Feng, F. W. Heinemann, J. M. O'Connor and K. Meyer, J. Am. Chem. Soc., 2008, 130, 2806–2816 CrossRef CAS PubMed.
- O. P. Lam, C. Anthon, F. W. Heinemann, J. M. O'Connor and K. Meyer, J. Am. Chem. Soc., 2008, 130, 6567–6576 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic and experimental details, spectroscopic data for 1H NMR, SQUID and UV/vis and detailed X-ray crystallographic data in CIF format. CCDC 965196–965199. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc52799a |
|
| This journal is © The Royal Society of Chemistry 2014 |
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a