 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic super-electron-donors: initiators in transition metal-free haloarene–arene coupling†
Shengze
Zhou
a,
Greg M.
Anderson
a,
Bhaskar
Mondal
a,
Eswararao
Doni
a,
Vicki
Ironmonger
b,
Michael
Kranz
b,
Tell
Tuttle
*a and
John A.
Murphy
*a
aWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: tell.tuttle@strath.ac.uk; john.murphy@strath.ac.uk
bGlaxoSmithKline Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, UK
First published on 9th October 2013
Abstract
Recent papers report transition metal-free couplings of haloarenes to arenes to form biaryls, triggered by alkali metal tert-butoxides in the presence of various additives. These reactions proceed through radical intermediates, but understanding the origin of the radicals has been problematic. Electron transfer from a complex formed from potassium tert-butoxide with additives, such as phenanthroline, has been suggested to initiate the radical process. However, our computational results encouraged us to search for alternatives. We report that heterocycle-derived organic electron donors achieve the coupling reactions and these donors can form in situ in the above cases. We show that an electron transfer route can operate either with phenanthrolines as additives or using pyridine as solvent, and we propose new heterocyclic structures for the respective electron donors involved in these cases. In the absence of additives, the coupling reactions are still successful, although more sluggish, and in those cases benzynes are proposed to play crucial roles in the initiation process.
Results
An explosion of interest has arisen in dehalogenative couplings between haloarenes and arenes to form biphenyls under transition metal-free conditions1–28 since the original report of coupling of iodobenzene with pyridine and pyrazine by Itami in 2008.1 The excitement is easily understood, since these reactions avoid the use of costly complexes of palladium, which are the normal catalysts. A common feature of the recent reactions is the use of sodium or potassium tert-butoxide as base. The absence of any significant contamination with transition metals like palladium,1,2 together with the properties of the new coupling reactions, has led to a general consensus that the reactions proceed by aryl radical intermediates.1,6 The corollary coupling between haloarenes and styrenes, also triggered by alkali metal butoxides,3,7,12 likely follows the same route.The general case for a radical mechanism was well summarised in an essay by Studer and Curran9 and an attractive proposal from that source is shown in Fig. 1A. Here, the aryl radical 2 adds to benzene to form the arylcyclohexadienyl radical 3. Deprotonation by butoxide affords the radical anion 4, which transfers an electron to aryl halide 1, thereby forming biaryl 5 together with a new aryl radical 2, ensuring the propagation of the chain. The outstanding question relates to how the initial radical 2 is generated, i.e. the initiation mechanism, and that is the subject of this paper.
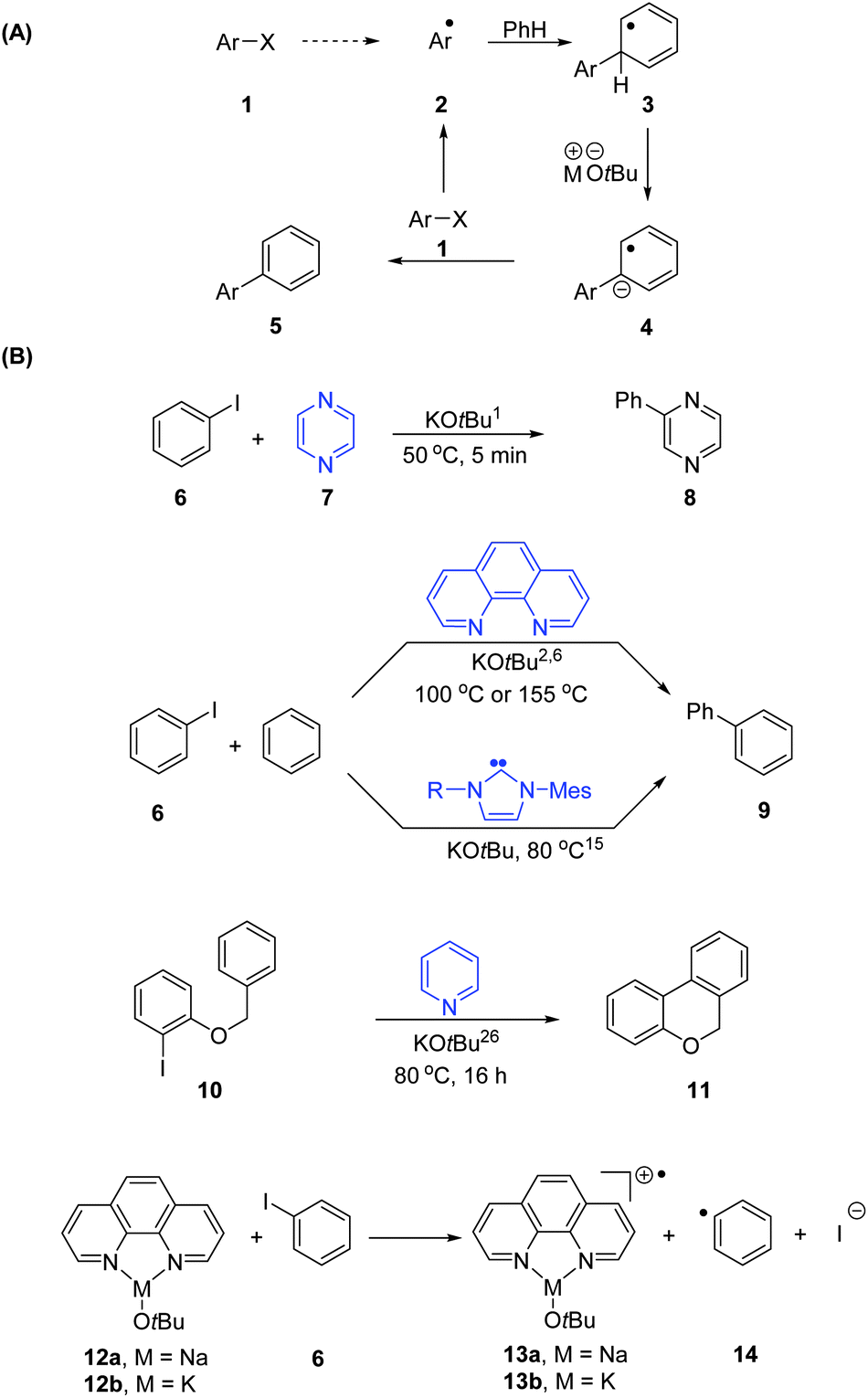 | ||
| Fig. 1 (A) Radical mechanism proposed by Studer and Curran.9 (B) Dehalogenative couplings between halobenzenes and arenes. | ||
With so many reports emerging in this area, our attention focused on four particular literature protocols that feature heterocycles as additives or substrates or solvent: (a) the Itami report1 showed arylation of pyridine and pyrazine 7 (Fig. 1B); (b) thorough investigations by Shi and coworkers2 and by Shirakawa, Hayashi et al.6 led to intermolecular arylation of benzenes to form biphenyl 9 and analogues in the presence of phenanthrolines; (c) the team of Chen and Ong15 showed similar success in the presence of N-heterocyclic carbenes, while (d) Charette26 led the breakthrough in discovering that cyclisations, for example of substrate 10, were significantly assisted by pyridine as solvent. Other additives have also proved useful in coupling reactions by other research groups, such as diamines,5,25 proline,13,21 pyridinium carboxylates,13 porphyrin,19 diols,5,23 alcohols,28 a macrocyclic pentapyridone,24 an unusual heterocycle11 and MOFs.14 Although iodoarenes are used most widely in the studies, bromoarenes2–7,13,14,18–20,24,26–28 and chloroarenes5–7,18,23,27 have also been often used.
A number of authors have suggested that electron transfer from a complex of NaOtBu or KOtBu with a ‘ligand’ would lead to electron transfer to an aryl halide like iodobenzene.2,3,6 Our starting point was to examine the thermodynamics for the basic electron transfer reaction between a phenanthroline complex of sodium tert-butoxide 12a and iodobenzene 6 as presented in the literature.6 The free energy change for formation of the products 13a and 14 and iodide ion from 12a and 6 is ΔG = +63.9 kcal mol−1, when calculated at the M06L/6-311G(d,p) level of theory using the CPCM continuum solvation method to incorporate the polarizing effect of the benzene solvent. The corresponding value for the analogous potassium case is ΔG = +59.5 kcal mol−1 (see ESI file for details†). These numbers were sufficiently large to give us grave concerns about radical initiation by this method. Of course, other unknown complexes of alkali metal tert-butoxides, for example involving aggregates rather than a single metal alkoxide, may be associated with less unfavourable thermodynamics for electron transfer, but the calculated values were sufficiently endergonic that they suggest other role(s) for the heterocycles that are highlighted in blue in Fig. 1B. Our findings apply to ground-state chemistry; more recently, Rossi et al. have reported photoactivation in the coupling of haloarenes to arenes.27 Photoactivation can achieve remarkable transformations, and we await further information on the mechanisms of those transformations.
We have recently shown29–34 that aryl iodides can be activated to form aryl radicals, and even aryl anions, in the presence of organic electron donors derived from pyridines and imidazoles. This raised an initial question of whether our electron donors could trigger haloarene–arene coupling reactions. Imidazolium salts 1529,30,32,35,36 can be deprotonated with strong bases like sodium hydride to form imidazolylidenes 16 (Fig. 2). Reaction between an imidazolylidene and an imidazolium salt 15 leads to C–C bond formation, and deprotonation of the intermediate by NaH affords intensely yellow electron donors 18. In like manner, pyridinium salts e.g., 19 were converted into bipyridinylidenes 20, which are strong electron donors that can reduce halobenzenes.31 However, the formation of these electron donors had not been demonstrated with metal butoxides as base nor in benzene as solvent. To this end, N-isopentylbenzimidazolium salt 21 was treated with potassium tert-butoxide in benzene, immediately affording the vibrant yellow solution characteristic of the bibenzimidazolylidene donor 22. In this case, 21 was selected rather than our previously used salt 23,29 because of our finding that the solubility of 24 was not high in benzene. As will be seen below in later experiments, where lower concentrations of disalt were employed, 23 was used then with no difficulty.
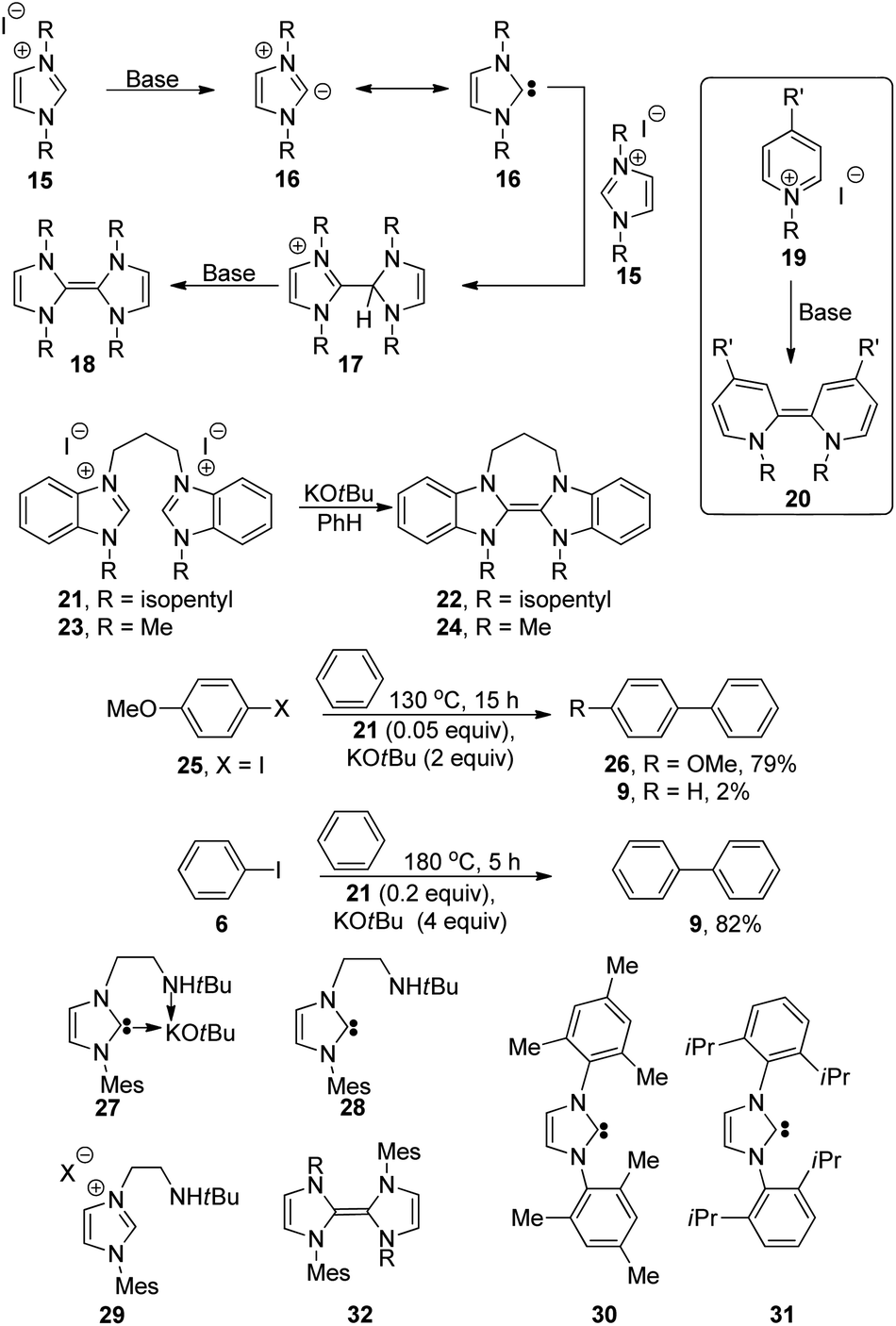 | ||
| Fig. 2 N-Heterocyclic carbenes and derived electron donors. | ||
Super-electron-donors (derived from 21 and 23) as initiators, and the relevance to N-heterocyclic carbenes as additives
Test reactions were then performed in benzene as solvent with salt 21 and KOtBu, using (a) p-iodoanisole 25 and (b) iodobenzene 6 as substrates, to explore possible coupling with benzene. Heating p-iodoanisole 25 with 21 (5 mol%) and KOtBu (2 equiv.) in benzene at 130 °C for 15 h afforded the desired 4-methoxybiphenyl 26 (79%), together with biphenyl 9 (2%). [Similar observations of small amounts of biphenyl in transition metal-free coupling reactions of substituted iodobenzenes with benzene have been made by Kappe and coworkers17 and will be discussed below.] Likewise, heating iodobenzene 6 at 180 °C for 5 h with 21 (20 mol%) and KOtBu (4 equiv.) in benzene and 130 °C for 3.5 h afforded biphenyl, 9, in very good yield (82% and 72% respectively). [Whereas these conditions were effective for coupling iodoarenes, bromobenzenes reacted very sluggishly. Thus reacting 4-bromotoluene with 23 (20 mol%) and KOtBu in benzene at 185 °C for 2.5 h afforded 4-methyl-1,1′-biphenyl (3%, not shown in scheme). This lower reactivity with aryl bromides mirrored our prior reactions of benzimidazole-derived donors29 with halobenzenes.]These findings are consistent with our previous chemistry of neutral organic electron donors. Our results may have particular relevance to the findings of Chen and Ong,15 who proposed that a complex 27 (Mes = 2,4,6-trimethylphenyl) between potassium butoxide and N-heterocyclic carbene 28, which derives from imidazolium salt 29 on treatment with KOtBu, could undergo electron transfer to an iodobenzene as part of the reaction mechanism for its coupling to benzene. They reported that carbenes 30 and 31, substituted by two bulky substituents led to very poor yields, while carbene 28 bearing one bulky substituent and one much less bulky substituent, operated very successfully. They attributed this success to the fact that the less bulky substituent in 28 featured an amine unit that might help to complex the metal, forming 27 and facilitating electron transfer, but an alternative interpretation would be that this substituent permitted the formation of the recognized super-electron-donor skeleton within 32 (R = tBuNHCH2CH2); formation of analogous structures from 30, 31 would be much more difficult for steric reasons.
Studer and Curran9 had proposed that radical generation might be required simply to initiate the reaction and that a chain reaction might then be sustained through electron transfer from an arene radical anion 4. In line with this, iodobenzene was heated at 180 °C in benzene for 6 h with KOtBu (3 equiv.) and decreasing amounts of salt 23 (0.1 equiv., 0.05 equiv. and 0.01 equiv.); biphenyl 9 was still formed in 65%, 67% and 73% yield respectively, consistent with the proposal that these electron donors are important simply for initiation of the radical process.
The role of temperature in these reactions was next investigated. At 130 °C for 3.5 h, in the presence of salt 21 (5 mol%), and KOtBu (2 equiv.), biphenyl product 9 (80%) was still formed. Dropping the temperature to 110 °C and keeping the concentration of the salt and base constant afforded biphenyl 9 (47%) together with recovered iodobenzene 6 (26%). Repeating this last experiment but dropping the concentration of salt 21 to 2.5 mol% and keeping the duration at 3.5 h still afforded product 9 (27%). These last experiments were then compared with ‘blank’ experiments at 130 °C and 110 °C respectively, i.e. where no salt 21 was present. In these cases, biphenyl 9 was still isolated in 30% and 27% yield respectively, indicating that the reaction occurs even in the absence of salt 21. Thus, at the lowest temperature, 110 °C, a comparison of the yields (27% in the presence of 2.5 mol% salt 21 and 27% in its absence) indicates that salt 21 is ineffective at assisting the reaction. In our previous work in DMF/toluene,29 our related electron donor 24 needed thermal activation to accomplish dehalogenation of arenes, and the threshold here for donor 22 appears to be in the 110–130 °C range.
To see if the reaction would become more efficient at higher temperature in the absence of salt 21, biphenyl 9 was isolated in 48% yield when iodobenzene 6 and KOtBu (4 equiv.) were heated at 185 °C for 14 h.
Initiation in the presence of KOtBu, but no additional additives
In these “additive-free” cases, only KOtBu, iodobenzene and benzene were present. Given the thermodynamic barrier for electron transfer mentioned above, butoxide cannot be the direct initiator of the radical chemistry, and this suggested that radicals could be formed in another way. Literature shows that potassium tert-butoxide can convert iodobenzene to benzyne 33 (Fig. 3A).37 Indeed, evidence for benzyne formation is cited in a number of the current publications relating to haloarene–arene coupling,6,17 although the regiochemistry of the observed products of haloarene–arene coupling, and other properties of the coupling reactions, mean that the isolated biphenyls do not derive from benzynes in any appreciable measure. We envisage that a very small concentration of benzyne could be formed, as part of the initiation process solely. The process is supported by the calculated energetics of the reaction (Fig. 3B). The abstraction of a proton and subsequent formation of KI is calculated to be an endergonic reaction (ΔG = 18.6 kcal mol−1). The products and intermediates of this mechanism were characterized as true minima on the potential energy surface and connected to the corresponding transition states. However, the inclusion of thermal and entropic effects results in the reverse reaction (33 → 6) occurring in a barrierless process and as such only a very low concentration of 33 will be present in situ. | ||
| Fig. 3 (A) Benzyne reacts with pyridine and base to form organic electron donors. (B) Reaction free energy (ΔG) profile for the formation of 33 from 6. (C) Reaction free energy (ΔG) profile for the formation of 36 from pyridine and 33. (D) Reaction free energy (ΔG) profile for the formation of 35 from 34. (E) Reaction free energy (ΔG) profile for the formation of 38 from the cation 35* and 36. ΔG values are reported in kcal mol−1. | ||
The literature also shows that benzyne is attacked by pyridines as nucleophiles.38 With this in mind, we envisaged a scenario where pyridine, under Charette's conditions,26 and possibly phenanthroline, under the conditions of Shi2 and of Shirakawa and Hayashi6 could similarly add to benzyne. In the case of pyridine, (Fig. 3) this would create a zwitterion 34 that could undergo proton transfer intramolecularly to form 36 (see Fig. 3C). Alternatively tert-butanol could protonate 34 (Fig. 3D), and the resulting salt 35 would then closely resemble the pyridinium salts 19 that were used by us previously as precursors of strong bipyridinylidene electron donors 20. Deprotonation by KOtBu could then lead to 36. The addition of the cation 35* to 36 results in the transformation to electron donors 38 and 40 (Fig. 3E). The rate-limiting step in the reaction is the formation of benzyne (33), which is thermodynamically unfavourable. However, once produced, the reaction to form the electron donor 38 is both kinetically and thermodynamically favourable. The formations of the various intermediates are all exergonic with the largest barrier encountered for the intramolecular proton transfer to form 36 from 34 (ΔG* = 9.3 kcal mol−1, Fig. 3C), which is easily accessible under the reaction conditions. To test this idea, N-phenylpyridinium chloride 35* (X = Cl) (0.2 equiv.) was treated with iodobenzene 6 and KOtBu (4 equiv.) in benzene at 180 °C for 8 h. This did indeed afford a good yield of biphenyl 9 (68%) showing that pyridinylidenes like 36 are possible intermediates in these reactions. Hence the reaction might be operating with benzyne as an intermediate. This would be attacked by pyridine (and possibly phenanthroline), ultimately resulting in the synthesis of bipyridinylidene (biphenanthrolylidene) super electron donors to initiate the radical chemistry.
However, the results that were seen above in the “additive-free” cases i.e., simply with iodobenzene, benzene and KOtBu still require an explanation of how these reactions can lead to radical chemistry. Benzyne reacts with benzene to form biphenyl as well as other products.40 Although detailed mechanistic studies of this reaction have not been carried out, the likely mechanism can be deduced from the reaction of benzyne with alkenes and thiones. Benzynes undergo reactions with alkenes to form benzocyclobutanes. At least two detailed studies of benzocyclobutane formation on reaction of benzyne with alkenes have shown that this is not a concerted reaction, but proceeds through a diradical intermediate.41,42 Similarly, addition to thiones affords diradical intermediates.43,44 Applying a similar reactivity to addition of benzyne to benzene would afford a diradical 41 (Fig. 4), and its similarity to phenylcyclohexadienyl radical 3 is apparent. Further reaction of the aryl radical in 41, by hydrogen atom abstraction from benzene (see below), followed by deprotonation of the cyclohexadienyl radical by base is a likely sequence to afford species, 43, that can act as an excellent electron-donor. As in the case of the reaction of benzyne with pyridine, the formation of the benzyne is the limiting step in this reaction. The calculated energetics show that the electron donor 43 will be formed in an exergonic reaction (ΔG = −29.1 kcal mol−1) with a maximum barrier of 12.2 kcal mol−1, once the reactive benzyne species is formed (see Fig. S2 in ESI†).
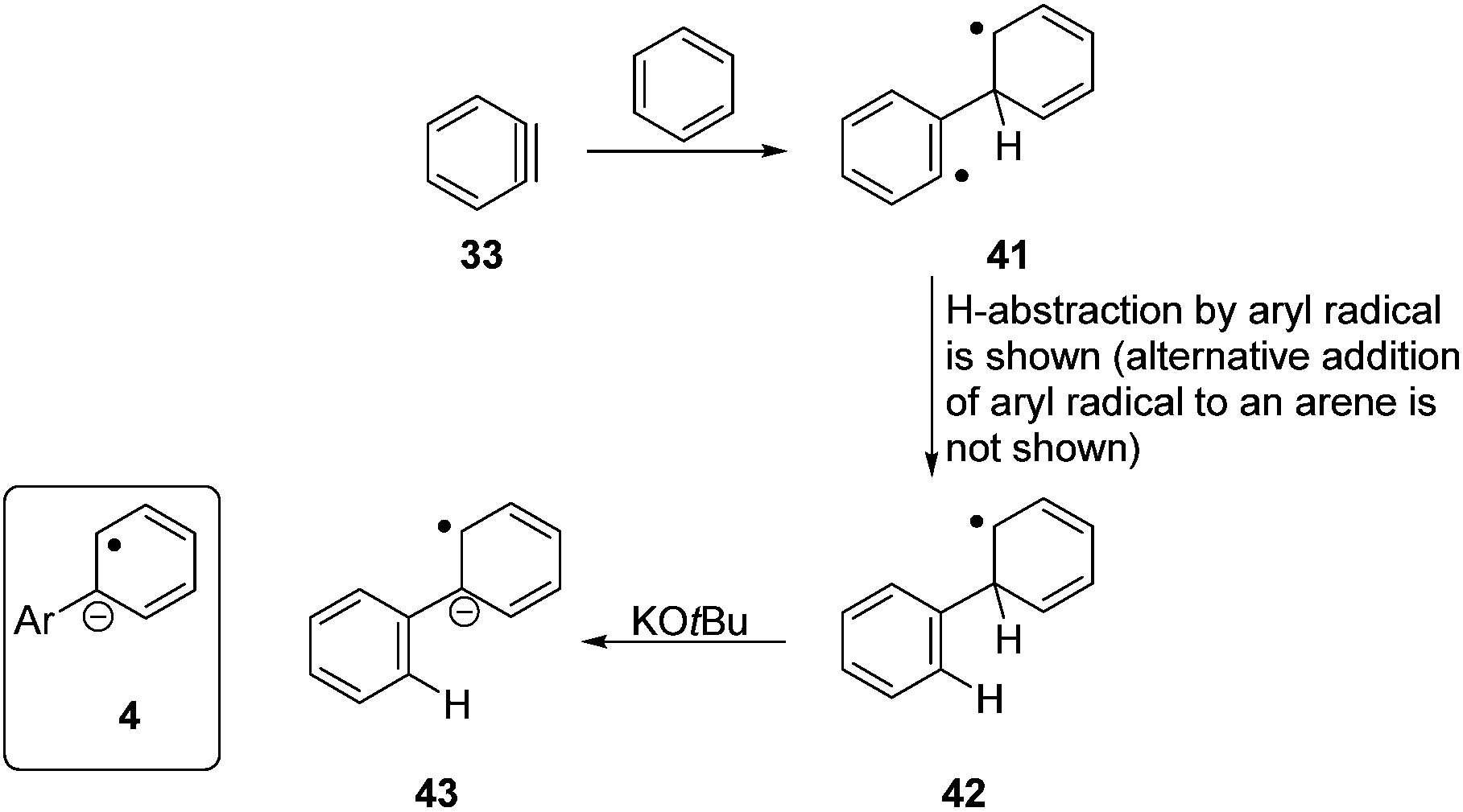 | ||
| Fig. 4 Proposed pathway from benzyne to an electron donor in the absence of “additives”. | ||
Our aim was now to focus on the role of electron transfer, in experiments that would preclude the formation of benzyne. Plainly, benzyne cannot form from iodobenzenes that are blocked in both ortho-positions, so 2,6-dimethyliodobenzene 44 was selected (Fig. 5) and treated with KOtBu and benzene (130 °C, 22 h). This led to no reaction (cf. the “additive-free” reaction above using iodobenzene 6). This experiment supports a role for benzyne under these “additive-free” conditions, for substrates like iodobenzene where benzyne formation is possible. The lack of reaction with 44 also showed that under these conditions KOtBu was not able to trigger electron transfer. It might have been argued that 2,6-dimethyliodobenzene 44 was too hindered to undergo electron transfer-mediated loss of iodide to form radical 47, but this argument was quashed when it was reacted with the benzimidazolium salt 21 (20 mol%) in the presence of KOtBu and benzene (130 °C, 22 h). This led to an inseparable mixture of 2,6-dimethylbiphenyl 46 (5%) and biphenyl 9 (19%) as well as recovery of 44 (36%). This suggests that the 2,6-dimethylphenyl radical 47 is not an efficient coupling agent, which is understandable for steric reasons, but also shows that this radical can abstract a hydrogen atom from the solvent, benzene, to form a phenyl radical 48,45 explaining the origin of the biphenyl 9 here and also from p-iodoanisole 25 reported above and also in the literature.17
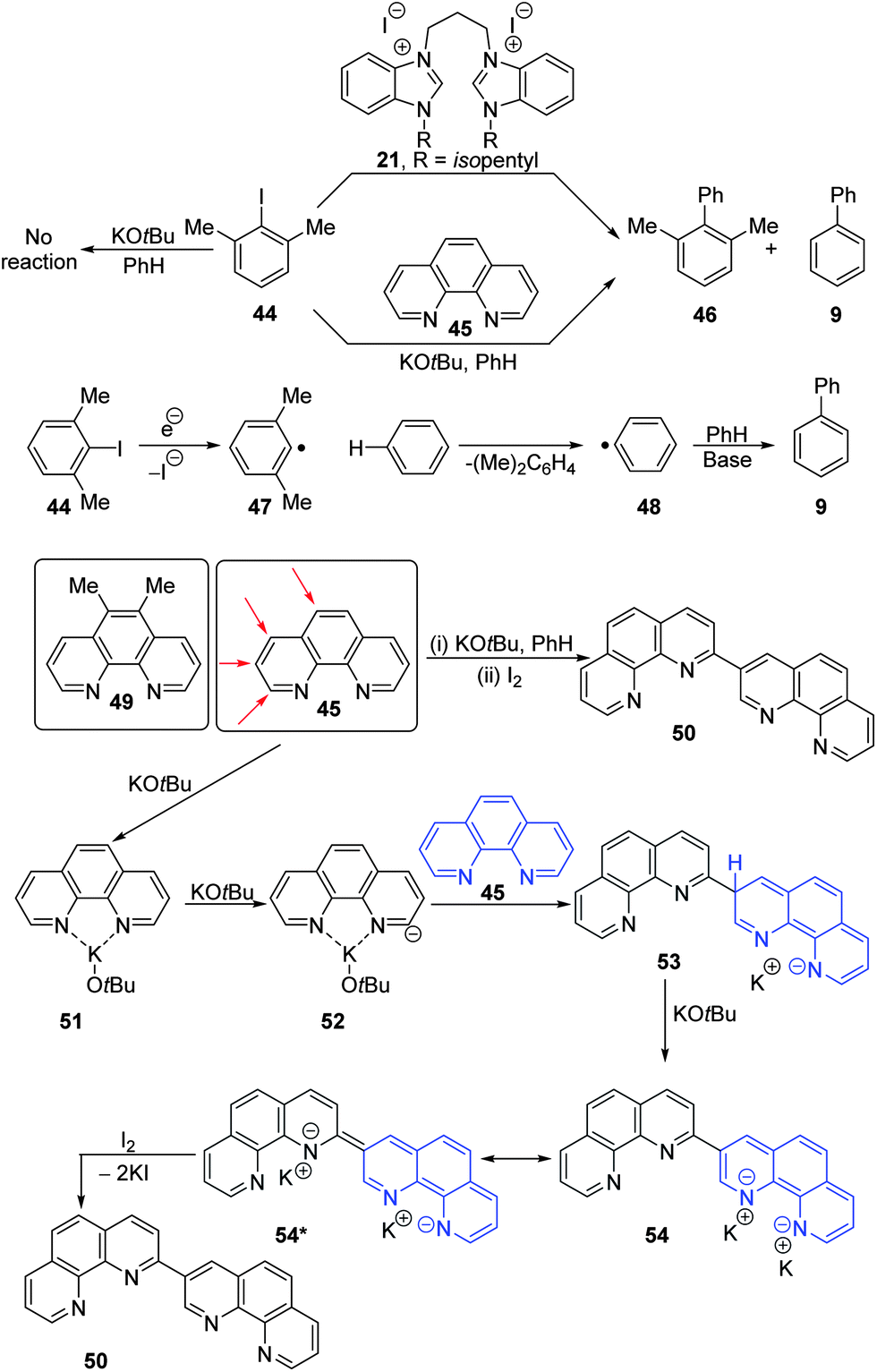 | ||
| Fig. 5 Reactivity of 2,6-dimethyliodobenzene with organic electron donors. | ||
It was now of interest to revisit additives, such as phenanthroline and pyridine. If they could convert substrate 44 to coupled product, then this would be evidence in favour of an electron-transfer capability in their reactions. Phenanthrolines were examined first.
Initiation with phenanthrolines as additives
Reaction of substrate 44 with phenanthroline 45 (20 mol%), KOtBu (2 equiv.) at 130 °C for 18 h afforded an inseparable mixture of biphenyl 9 (19%) and 2,6-dimethylbiphenyl 46 (5%) in the same yield and ratio as had been seen with the salt 21, precursor of the organic electron donor 22, supporting an electron transfer mechanism for reactions involving phenanthroline 45. Also recovered was 44 (41%).Next, 5,6-dimethylphenanthroline 49 (100 mol%) was reacted. Complete conversion was seen after heating at 130° C for 15 h, affording the same mixture of biphenyl 9 (27%) and 2,6-dimethylbiphenyl 46 (8%). Repeating the experiment with dimethylphenanthroline 49 (20 mol%) led to biphenyl 9 (17%) and 2,6-dimethylbiphenyl 46 (5%), together with recovered 44 (31%). In these reactions with phenanthrolines, we noted that significant quantities of a deep green solid material were produced during the reactions, and this was now investigated.
Phenanthroline2–4,6–845 is an electron-poor arene, and an obvious way to convert it into a π-electron-rich species would be by addition of a nucleophile to its periphery. We considered the possibilities shown in Fig. 5; addition of a nucleophile at any of the atoms indicated by the red arrows on boxed structure 45 would afford electron-rich anionic adducts that ultimately might convert into electron donors resembling those already discussed in this paper. To probe this, phenanthroline was heated in benzene in the presence of potassium tert-butoxide. As in the previous reactions, deep-green solid material was produced, and at the end of the reaction, this was analysed. When removed from the glove-box and exposed to air, this material was pyrophoric, reminiscent of our previous organic electron donors. When the material was instead quenched with iodine, as electron acceptor, and was then examined by 1H NMR, it appeared to be an almost pure single compound. Rigorous purification through column chromatography gave pure (2,3′-bis)phenanthroline 50 as a red oil (36%); a key feature of its spectrum was the presence at δ 9.42 and δ 9.84 of two doublets each with J = 2.2 Hz, indicating meta coupling. To rationalize its formation, complexation of phenanthroline with a potassium ion would give structure 51 (KOtBu is represented as monomeric here, but we recognize that aggregation may occur in benzene). Some of these molecules are then deprotonated to form 52. This anion then acts as a nucleophile on the 3-position of another phenanthroline molecule to afford intermediate 53. Further deprotonation by KOtBu would afford the dianion 54, with the charge highly delocalized over the six rings, and undoubtedly complexed to potassium. The analogy is clear between canonical form 54 and super-electron donors 20 and 38, all of which feature very electron-rich nitrogen heterocycles, where loss of two electrons restores full aromaticity in all three cases. The energetics of this reaction mechanism were investigated computationally (see Fig. S3 in the ESI†). The rate-limiting step in the reaction was found to be the initial ortho deprotonation of phenanthroline (51) to form 52. This reaction is endergonic by 15.8 kcal mol−1, however in analogy to the formation of benzyne, the reverse reaction from 52 to form 51 is barrierless and as such only very small amounts of 52 are present in situ. However, once 52 is formed, a subsequent reaction with phenanthroline can readily occur. The maximum barrier (ΔG*) for the addition to the meta position of phenanthroline is 10.5 kcal mol−1. This is slightly more favourable than the addition to the para (10.6 kcal mol−1) or ortho (11.5 kcal mol−1) positions. Overall, the formation of 54 is strongly favourable with a reaction energy of −30 kcal mol−1.
Since phenanthroline had converted substrate 44 to products 46 and 9, pyridine (1 equiv.) was also reacted with KOtBu (2 equiv.) at 130 °C for 15 h. However, no reaction was observed.
Initiation with pyridine as solvent and relevance to pyridines or pyrazines as substrates
The lack of reactivity of pyridine is noteworthy, at least under these conditions where it is present at this limited concentration. Under the Charette conditions,26 where pyridine has been shown to facilitate intramolecular coupling reactions, pyridine is present in great excess as the solvent, and so substrate 55a was now tested under these conditions (Fig. 6). The 2-benzyloxyiodobenzene 55a underwent complete conversion and afforded an inseparable mixture of two products, 56a (56%) and 57a (18%), identified by NMR and mass spectrometry. Both of these products are consistent with intermediate aryl radicals; product 56a is the expected cyclisation product from the Charette conditions,26 and 57a should result from quenching of the intermediate aryl radical by hydrogen abstraction from pyridine, analogous to the reaction of aryl radical 47 with benzene shown in Fig. 5. The studies were then extended to the 2,6-dibenzyloxyiodobenzene 55b. This behaved in a qualitatively similar way, affording an inseparable mixture of two products, 56b (9%) and 57b (15%).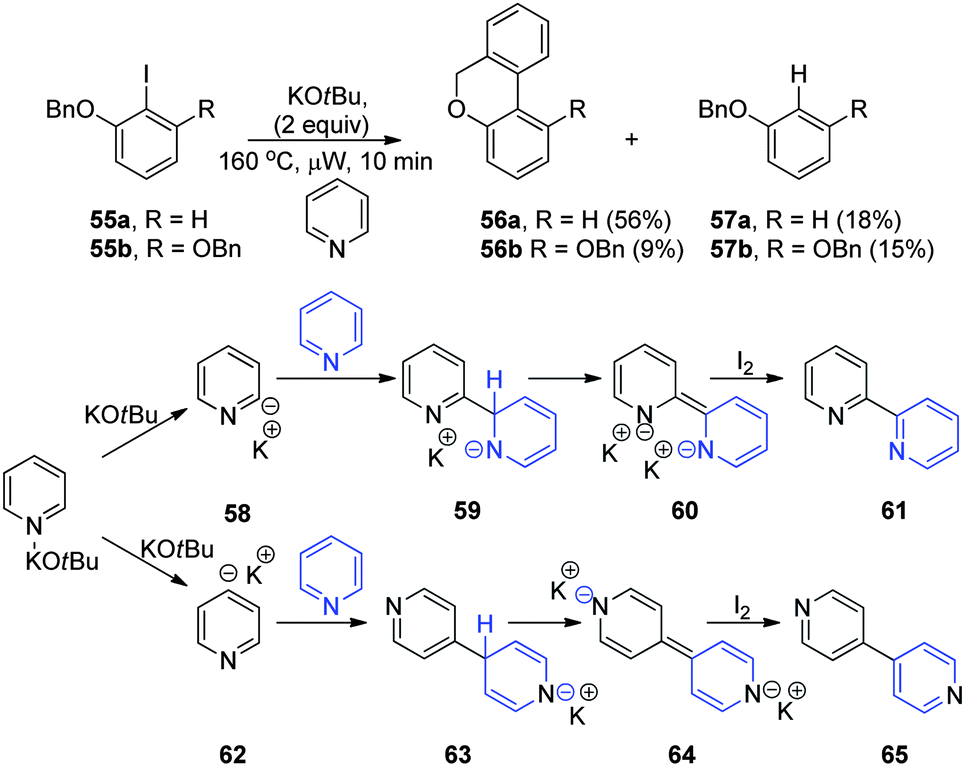 | ||
| Fig. 6 Base-induced formation of organic electron donors from pyridine. | ||
Under these conditions, and with this substrate 55b, benzyne cannot be an intermediate, and so initiation results from electron transfer. Based on the phenanthroline studies above, it appeared likely that a similar reaction occurs to form an organic electron donor. Pyridine may be much more difficult to deprotonate than phenanthroline, since the complex between pyridine and KOtBu should be a lot weaker than the complex between the chelating phenanthroline and KOtBu; hence lower concentrations of pyridine-derived super electron donor would be formed. A blank experiment was again conducted in which potassium tert-butoxide was heated in pyridine as solvent. On work-up with iodine as electron acceptor, this led to very small quantities of a residue. This residue was initially examined by GCMS and gave two peaks that showed the correct m/z for (two isomers of) bipyridine. Purification provided mass spectra and 1H NMR spectra indicating 2,2′-bipyridine 61 (the major product) and 4,4′-bipyridine 65 (no 2,4′-bipyridine was isolated). The origin of 61 is not difficult to rationalize: pyridine is deprotonated in the 2-position to form 58. Addition of 58 to the electrophilic 2-position of pyridine would afford intermediate 59 that, on further deprotonation by KOtBu, would afford dianions 60. Finally, oxidation with iodine affords bipyridine 61. Similarly, 62 resulting from deprotonation of pyridine in the 4-position39 can lead to 65. Compounds 60 and 64 again contain the signature properties of a super-electron-donor, i.e. a very electron-rich nitrogen-containing heterocyclic structure that can become completely aromatic by loss of two electrons.
These reactions relate to the use of pyridine as solvent, as seen in the work of Charette's team,26 but also to the initial observations of Itami,1 where pyridines and pyrazines were reacted in the presence of iodoarenes and KOtBu.
Conclusions
In summary, we propose that initiation of coupling reactions between haloarenes and arenes in the absence of transition metals can occur, based on formation of heterocycle-derived organic electron donors in situ by different pathways. Importantly, they avoid the requirement for KOtBu, or a simple derived complex, to act as an electron donor. Our findings provide a rationale, based on the developing chemistry of organic super electron donors, for the generation of radicals to initiate transition metal-free coupling reactions under four different protocols; here, the heterocycles do not form organocatalysts, but rather, organic initiators, whose reactions add to the growing litany of reactions that are accomplished by organic electron donors. A final interesting point is that the coupling of simple iodobenzenes and benzene proceeds also in the absence of additives – here a role for benzyne in the initiation of the radical chemistry is proposed.Acknowledgements
We thank ORSAS, EPSRC, GSK and University of Strathclyde for funding. Mass spectrometry data were acquired at the EPSRC UK National Mass Spectrometry Facility at Swansea University.Notes and references
- S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673–4676 CrossRef CAS PubMed.
- C. L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, Z.-B. Li and J. Shi, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed.
- C. L. Sun, Y.-F. Gu, B. Wang and Z.-J. Shi, Chem.–Eur. J., 2011, 17, 10844–10847 CrossRef CAS PubMed.
- C.-L. Sun, Y.-F. Gu, W.-P. Huang and Z.-J. Shi, Chem. Commun., 2011, 47, 9813–9815 RSC.
- W. Liu, H. Cao, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef CAS PubMed.
- E. Shirakawa, K.-I. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537–15539 CrossRef CAS PubMed.
- E. Shirakawa, X. Zhang and T. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 4671–4674 CrossRef CAS PubMed.
- E. Shirakawa and T. Hayashi, Chem. Lett., 2012, 41, 130–134 CrossRef CAS.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018–5022 CrossRef CAS PubMed.
- N. E. Leadbeater, Nat. Chem., 2010, 2, 1007–1009 CrossRef CAS PubMed.
- G.-P. Yong, W.-L. She, Y.-M. Zhang and Y.-Z. Li, Chem. Commun., 2011, 47, 11766–11768 RSC.
- M. Rueping, M. Leiendecker, A. Das, T. Poisson and L. Bui, Chem. Commun., 2011, 47, 10629–10631 RSC.
- Y. Qiu, Y. Liu, K. Yang, W. Hong, Z. Li, Z. Wang, Z. Yao and S. Jiang, Org. Lett., 2011, 13, 3556–3559 CrossRef CAS PubMed.
- H. Liu, B. Yin, G. Z. Gao, Y. Li and H. Jiang, Chem. Commun., 2012, 48, 2033–2035 RSC.
- W.-C. Chen, Y.-C. Hsu, W.-C. Shih, C.-Y. Lee, W.-H. Chuang, Y.-F. Tsai, P. P.-Y. Chen and T.-G. Ong, Chem. Commun., 2012, 48, 6702–6704 RSC.
- H. Zhang, R. Shi, A. Ding, L. Lu, B. Chen and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 12542–12545 CrossRef CAS PubMed.
- B. Pieber, D. Cantillo and O. C. Kappe, Chem.–Eur. J., 2012, 18, 5047–5055 CrossRef CAS PubMed.
- B. S. Bhakuni, A. Kumar, S. J. Balkrishna, J. A. Sheikh, S. Konar and S. Kumar, Org. Lett., 2012, 14, 2838–2841 CrossRef CAS PubMed.
- Y. S. Ng, C. S. Chan and K. S. Chan, Tetrahedron Lett., 2012, 53, 3911–3914 CrossRef CAS.
- S. De, S. Ghosh, S. Bhunia, J. A. Sheikh and A. Bisai, Org. Lett., 2012, 14, 4466–4469 CrossRef CAS PubMed.
- K. Tanimoro, M. Ueno, K. Takeda, M. Kirihata and S. Tanimori, J. Org. Chem., 2012, 77, 7844–7849 CrossRef CAS PubMed.
- L. G. Wang, G. B. Yan and X. Y. Zhang, Chin. J. Org. Chem., 2012, 32, 1864–1871 CrossRef CAS.
- Y. Wu, S. M. Wong, F. Mao, T. L. Chan and F. Y. Kwong, Org. Lett., 2012, 14, 5306–5309 CrossRef CAS PubMed.
- H. Zhao, J. Shen, J. Guo, R. Ye and H. Zeng, Chem. Commun., 2013, 49, 2323–2325 RSC.
- I. Thome and C. Bolm, Org. Lett., 2012, 14, 1892–1895 CrossRef CAS PubMed.
- D. S. Roman, Y. Takahashi and A. B. Charette, Org. Lett., 2011, 13, 3242–3245 CrossRef CAS PubMed.
- M. E. Buden, J. F. Guastavino and R. A. Rossi, Org. Lett., 2013, 15, 1174–1176 CrossRef CAS PubMed.
- W. Liu, F. Tian, X. Wang, H. Yu and Y. Bi, Chem. Commun., 2013, 49, 2983–2985 RSC.
- J. A. Murphy, T. A. Khan, S.-Z. Zhou, D. W. Thomson and M. Mahesh, Angew. Chem., Int. Ed., 2005, 44, 1356–1360 CrossRef CAS PubMed.
- J. A. Murphy, S.-Z. Zhou, D. W. Thomson, F. Schoenebeck, M. Mohan, S. R. Park, T. Tuttle and L. E. A. Berlouis, Angew. Chem., Int. Ed., 2007, 46, 5178–5183 CrossRef CAS PubMed.
- J. A. Murphy, J. Garnier, S. R. Park, F. Schoenebeck, S.-Z. Zhou and A. T. Turner, Org. Lett., 2008, 10, 1227–1230 CrossRef CAS PubMed.
- P. I. Jolly, S. Zhou, D. W. Thomson, J. Garnier, J. M. Parkinson, T. Tuttle and J. A. Murphy, Chem. Sci., 2012, 3, 1675–1679 RSC.
- E. Doni, S. O'Sullivan and J. A. Murphy, Angew. Chem., Int. Ed., 2013, 52, 2239–2242 CrossRef CAS PubMed.
- S. Zhou, H. Farwaha and J. A. Murphy, Chimia, 2012, 66, 418–425 CrossRef CAS PubMed.
- T. A. Taton and P. Chen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1011–1013 CrossRef CAS.
- R. W. Alder, M. E. Blake, L. Chaker, J. N. Harvey, F. Paolini and J. Schütz, Angew. Chem., Int. Ed., 2004, 43, 5896–5911 CrossRef CAS PubMed.
- G. B. Bajracharya and O. Daugulis, Org. Lett., 2008, 10, 4625–4628 CrossRef CAS PubMed.
- M. Jeganmohan, S. Bhuvaneswari and C.-H. Cheng, Chem.–Asian J., 2010, 5, 153–159 CrossRef CAS PubMed.
- Deprotonation in the 4-position of pyridine may also occur; see: M. Albrecht and H. Stoeckli-Evans, Chem. Commun., 2005, 4705–4707 RSC.
- R. G. Miller and M. Stiles, J. Am. Chem. Soc., 1963, 85, 1798–1800 CrossRef CAS.
- P. G. Gassman and H. P. Benecke, Tetrahedron Lett., 1969, 10, 1089–1092 CrossRef.
- A. T. Bowne, T. A. Christopher and R. H. Levin, Tetrahedron Lett., 1976, 14, 4111–4114 CrossRef.
- S. Yamabe, T. Minato, A. Ishiwata, O. Irinamihira and T. Machiguchi, J. Org. Chem., 2007, 72, 2832–2841 CrossRef CAS PubMed.
- K. Okuma, S. Sonoda, Y. Koga and K. Shioji, J. Chem. Soc., Perkin Trans. 1, 1999, 2997–3000 RSC.
- X. Qian, P. Mao, W. Yao and X. Guo, Tetrahedron Lett., 2002, 43, 2995–2998 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, computational details and additional calculations and key spectra are provided. See DOI: 10.1039/c3sc52315b |
| This journal is © The Royal Society of Chemistry 2014 |