Photoredox activation and anion binding catalysis in the dual catalytic enantioselective synthesis of β-amino esters†
Giulia
Bergonzini
a,
Corinna S.
Schindler
b,
Carl-Johan
Wallentin
a,
Eric N.
Jacobsen
*b and
Corey R. J.
Stephenson
*a
aDepartment of Chemistry, University of Michigan, 930 N University Ave, Ann Arbor, USA. E-mail: crjsteph@umich.edu; Fax: +1 734-647-4865; Tel: +1-734-763-8283
bDepartment of Chemistry, Harvard University, Cambridge, USA. E-mail: Jacobsen@chemistry.harvard.edu; Fax: +1-617496-3688; Tel: +1-617-496-1880
First published on 23rd August 2013
Abstract
The enantioselective oxidative C–H functionalization of tetrahydroisoquinoline derivatives is achieved through the merger of photoredox and asymmetric anion-binding catalysis. This combination of two distinct catalysis concepts introduces a potentially general approach to asymmetric transformations in oxidative photocatalysis.
Introduction
Photoredox catalysis has emerged recently as a powerful tool for organic synthesis.1 Irradiation of photoactive catalysts with visible light can be parlayed to the practical generation of synthetically useful high-energy organic intermediates such as free radicals2 and radical anions and cations.3,4 Successful efforts to induce enantioselective catalytic control in such photocatalyzed processes have relied thus far on covalent organocatalysis: photoredox aminocatalysis as introduced by MacMillan,2a,5 and the combination of photoredox catalysis with N-heterocyclic carbene catalysis as developed by Rovis.6,7Since many reactions initiated by visible light photocatalysis involve the reductive generation of halide anions,8 we considered whether the use of chiral anion-binding catalysts9 in combination with photocatalysis would provide opportunities for new types of enantioselective transformations.10,11 In one specific embodiment of this idea, we envisioned that iminium ion equivalents generated under mild conditions from tertiary amines by photocatalyzed oxidation12 might be trapped in stereoselective nucleophilic addition reactions under the influence of a chiral H-bond donor catalyst (Fig. 1). We describe here the successful development of a dual catalyst strategy for the enantioselective oxidative alkylation of tetrahydroisoquinolines with silyl ketene acetals.
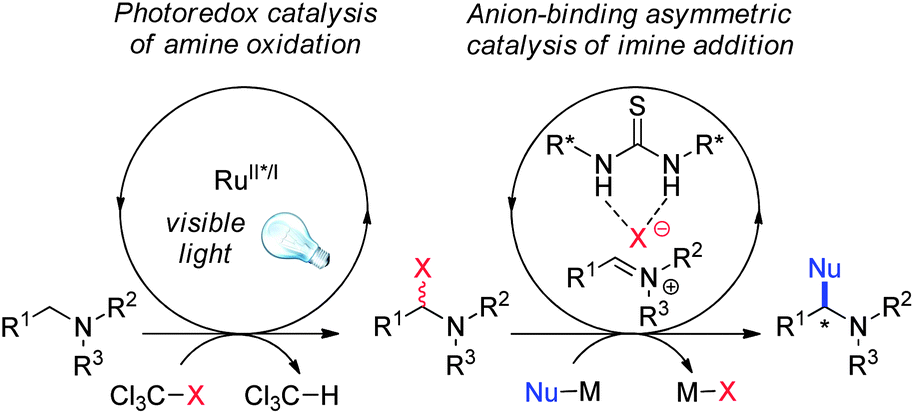 | ||
| Fig. 1 Merger of photoredox and anion-binding catalysis in oxidative enantioselective C–H functionalization of amines. | ||
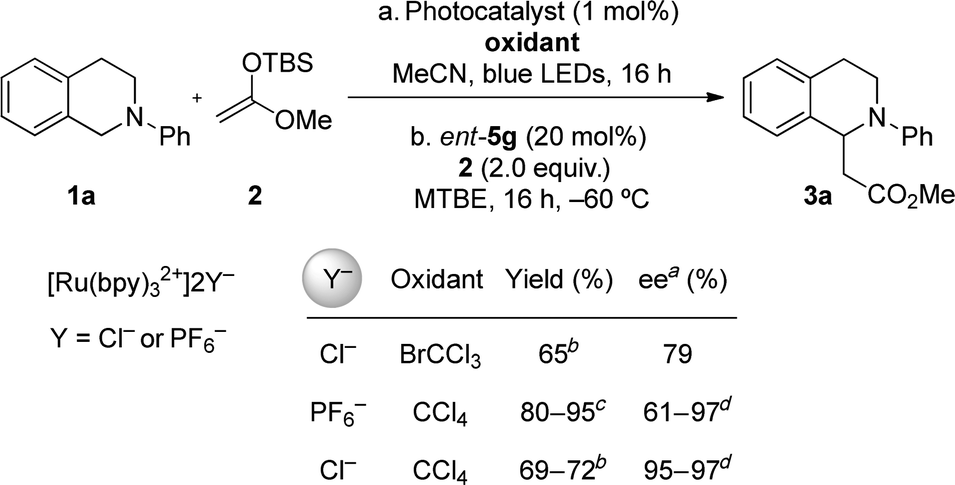
We chose to evaluate the proposed dual catalytic reaction design in enantioselective oxidative Mannich reactions to access tetrahydroisoquinoline derived β-amino esters such as 3a (Table 1).13,14 In this context, we reported recently a photocatalytic method for the oxidative generation of iminium ion precursors from simple N-aryltetrahydroisoquinolines such as 1a using bromotrichloromethane as the stoichiometric oxidant.15–17 Coupling this protocol to an enantioselective thiourea-catalyzed addition of enolate equivalents such as silyl ketene acetal 2 would require identification of the appropriate photoredox and chiral thiourea catalysts, as well as reaction conditions suitable for both transformations.
|
|
|||||
|---|---|---|---|---|---|
|
|
|||||
| Entry | Thiourea catalyst | T a (°C) | [1a]° (M) | Yieldb [%] | eec [%] |
| a Reaction temperature for the alkylation step in MTBE. b Yields determined by 1H NMR spectroscopic analysis of the crude reaction mixture relative to 2,5-dimethylfuran as the internal standard. c Determined by HPLC on commercial chiral columns. d Pyrrolidine configuration is (R), tert-leucine configuration is (R). e Pyrrolidine configuration is (R), tert-leucine configuration is (S). | |||||
| 1 | — | −78 | 0.1 | 0 | N/A |
| 2 | 4 | −78 | 0.1 | 75 | 10 |
| 3 | 5a | −78 | 0.1 | 68 | 50 |
| 4 | 5b | −78 | 0.1 | 63 | 20 |
| 5 | 5c | −78 | 0.1 | 57 | 80 |
| 6 | 5d | −78 | 0.1 | 63 | 29 |
| 7 | 5c | −60 | 0.05 | 37 | 93 |
| 8 | epi-5cd | −60 | 0.05 | 68 | 44 |
| 9 | 5e | −60 | 0.05 | 63 | 87 |
| 10 | 5f | −60 | 0.05 | 63 | 85 |
| 11 | ent-5ge | −60 | 0.05 | 72 | −97 |

Results and discussion
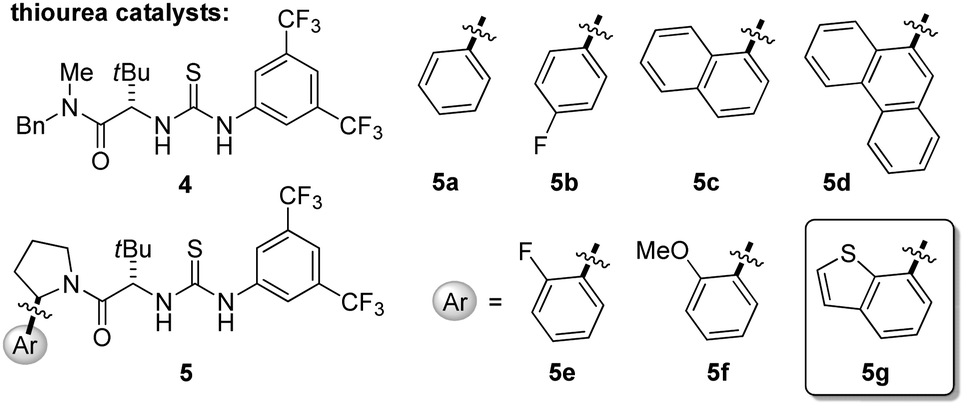
In initial experiments, the proper choice of solvent systems was revealed as a key challenge. In relatively polar media (CH3CN, CH2Cl2, DMF), with CCl4 as the stoichiometric oxidant, the oxidative photocatalytic Mannich reaction of N-phenyltetrahydroisoquinoline (1a) and silyl ketene acetal 2 afforded 3a cleanly in the presence of chiral thiourea catalysts, but always in racemic form. In contrast, unreacted tetrahydroisoquinoline 1a could be recovered from attempted oxidations in non-polar solvents such as MTBE, which are optimal for attaining high enantioselectivity in anion-binding thiourea catalysis. The failure of the photoredox reaction under these conditions could be ascribed simply to the lack of solubility of the photocatalyst, even when relatively non-polar complexes such as Ru(bpy)3(PF6)2 were employed. Ultimately, the enantioselective oxidative Mannich reaction could be achieved by performing the photocatalytic oxidation in CH3CN and switching solvents to MTBE for the thiourea-catalyzed alkylation reaction. In this manner, full conversion of 1a to the corresponding iminium ion in CH3CN was observed after irradiation with blue LEDs for 16 hours using 2 equiv of CCl4, and tetrahydroisoquinoline 3a was obtained in 50% ee upon alkylation with 2 in the presence of chiral thiourea 5a at −78 °C.Enantioselective oxidative Mannich reactions of 1a and silyl ketene acetal 2 were evaluated with a variety of thiourea catalysts (Table 1). Only catalysts bearing both 3,5-bis(trifluoromethyl)aniline and tertiary amide components were found to promote useful reaction rates at −78 °C.18 Constrained arylpyrrolidine-derived thiourea catalysts of the general structure 5, which have proven effective in a wide range of enantioselective transformations,19,20 induced significantly higher ee's than catalysts bearing a less constrained tertiary amide fragment (e.g.4). In that context, the relative configuration of the arylpyrrolidine unit relative to the tert-leucine component had a significant impact on reaction enantioselectivity (entries 7–8), pointing to the importance of the precise disposition of the aryl group relative to the thiourea anion-binding site. No straightforward correlation between the expanse of the α-aryl substituent on the pyrrolidine and reaction enantioselectivity was observed (Table 1, entries 3–6). In contrast, substitution on the ortho-position of the arylpyrrolidine resulted in significant improvements in enantioinduction, although the electronic properties of the substituent had little impact (entries 9–11). Further reaction optimization using catalyst 5c revealed a significant improvement in enantioselectivity when the reaction was conducted at −60 °C with an initial concentration of 0.05 M (entries 5 and 7). After extensive evaluation of different arylpyrrolidine-derived analogs of 5, thiourea 5g was identified as an optimal catalyst for the model transformation of 1a to 3a.
During the course of the optimization studies outlined above, we observed that the identities of both the photocatalyst counterion and the halogen atom source had significant effects on enantioselectivity in the thiourea-catalyzed silyl ketene acetal addition reaction (Table 2). Consistently lower enantioselectivity was observed when using BrCCl3 as the stoichiometric oxidant compared with CCl4, a result that is attributable to more favorable interactions of the chloride-bound thiourea catalyst in the enantioselectivity-determining transition structure.21 When Ru(bpy)3(PF6)2 was used as the photocatalyst in place of Ru(bpy)3Cl2 together with CCl4 as the stoichiometric oxidant, highly variable enantioselectivities were obtained (e.g. 61–97% ee with catalyst ent-5g). This unpredictable behavior associated with the use of the Ru(bpy)3(PF6)2 photocatalyst appears to be tied to the heterogeneous nature of the alkylation reaction mixture. Although only 2 mol% of PF6− is present in the reaction medium and the iminium ion intermediate is predominantly associated with chloride (Cl−/PF6− = ca. 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the PF6− may impart enhanced solubility properties to the iminium ion, and thereby enable the racemic, background reaction to a greater extent.
:1), the PF6− may impart enhanced solubility properties to the iminium ion, and thereby enable the racemic, background reaction to a greater extent.
| a Determined by HPLC on commercial chiral columns. b Isolated yield after purification by chromatography on silica gel. c Determined by 1H NMR spectroscopic analysis of the crude reaction mixture relative to 2,5-dimethylfuran as the internal standard. d Range of enantiomeric excess achieved over >10 runs. |
|---|
|
With optimized conditions in hand we next explored the substrate scope of the reaction (Fig. 2). The time required for full conversion of the N-aryltetrahydroisoquinoline derivatives 1 in the oxidation reaction was highly dependent on the electronic nature of the substrate, with electron-rich derivatives being more readily oxidized. To ensure complete conversion to the iminium ion, the amine was treated under the photochemical oxidation conditions for 16 h in all cases. The position of substituents on the N-aryl group influenced the enantioselectivity of the oxidative alkylation addition more profoundly than did their electronic properties. Thus, ortho-substituted substrates generally provided higher ee compared to para-substituted analogs (3lvs.3b; 3jvs.3d). In contrast, reaction enantioselectivities were very sensitive to the electronic nature of substituents on the tetrahydroisoquinoline ring, with electron-rich systems generally affording lower ee's and/or yields (e.g.3e, 3h, 3i).
 | ||
| Fig. 2 Substrate scope. | ||
In order to better establish the synthetic utility of this chiral tetrahydroisoquinoline synthetic methodology, we sought to develop a protocol for N-dearylation of the products to provide access to the more useful secondary amine derivatives.22 Electron-rich anilines have favorable redox properties that render them prone to N-aryl bond cleavage under oxidative conditions.23 Accordingly, compound 3l, which was generated with nearly perfect enantioselection in the oxidative Mannich reaction was chosen as a model substrate for dearylation studies. Application of standard oxidative protocols for PMP or PMB removal (CAN, PhI(TFA)2, or DDQ) led to rapid consumption of 3l but low yields of the desired secondary amine due to product decomposition via over-oxidation. We anticipated that the use of an oxidant more closely matched to the oxidation potential of the substrate would have a better chance of avoiding decomposition of the relatively sensitive secondary amine product. Evaluation of Fe(III)-based oxidants led to the identification of [Fe(bpy)3]3+, formed in situ upon treatment of [Fe(bpy)3](PF6)2 with CAN,24 as a highly effective oxidant for this transformation, leading to clean conversion to the desired amine 6. Comparison of the optical rotation of 6 with the reported value enabled the assignment of its absolute configuration. Acylation of 6 with acetyl chloride provided 7 (95% ee, 87% yield starting from 3l) with no significant compromise of enantiomeric purity.
 | (1) |
Conclusions
In summary, we have developed an approach to the oxidative, enantioselective C–H functionalization of tetrahydroisoquinoline derivatives. A mild method for the selective dearylation of products bearing N-o-anisyl groups was developed, thereby enabling further derivatization of the tetrahydroisoquinoline scaffold. Further applications of photoredox/chiral anion binding dual catalysis are anticipated and are the subject of current study.Acknowledgements
Financial support for this research from the NIH-NIGMS (PO1 GM69721 and R01-GM43214) to (ENJ and R01-GM096129 to CRJS), the Alfred P. Sloan Foundation, Amgen, Boehringer Ingelheim, Eli Lilly, and Novartis is gratefully acknowledged. GB is grateful to the Spanish Ministry of Education and Science (MEC) for a FPU predoctoral fellowship (AP2009-0950). Postdoctoral support from the Alexander von Humboldt Foundation (Feodor Lynen fellowship to CSS) and the Swedish Research Council (to CJW) is gratefully acknowledged.Notes and references
- For recent reviews, see: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (b) F. Teplý, Collect. Czech. Chem. Commun., 2011, 76, 859 CrossRef; (c) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617 CrossRef CAS PubMed; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- For selected examples, see: (a) D. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed; (b) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756 CrossRef CAS PubMed; (c) R. S. Andrews, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2010, 49, 7274 CrossRef CAS PubMed; (d) J. W. Tucker and C. R. J. Stephenson, Org. Lett., 2011, 13, 5468 CrossRef CAS PubMed; (e) M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951 CrossRef CAS PubMed; (f) J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed.
- For a recent review on accessing the chemistry of radical ions, see: M. A. Ischay and T. P. Yoon, Eur. J. Org. Chem., 2012, 3359 CrossRef CAS.
- For selected examples, see: (a) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886 CrossRef CAS PubMed; (b) M. A. Ischay, Z. Lu and T. P. Yoon, J. Am. Chem. Soc., 2010, 132, 8572 CrossRef CAS PubMed; (c) S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350 CrossRef CAS PubMed; (d) S. Maity, M. Zhu, R. S. Shinabery and N. Zheng, Angew. Chem., Int. Ed., 2012, 51, 222 CrossRef CAS PubMed.
- (a) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875 CrossRef CAS PubMed; (b) H.-W. Shih, M. N. Vander Wal, R. L. Grange and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 13600 CrossRef CAS PubMed.
- D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094 CrossRef CAS PubMed.
- These are distinct conceptually from enantioselective catalysis of photochemical transformations, as described by Bach, et al. For examples, see: (a) A. Bauer, F. Westkämper, S. Grimme and T. Bach, Nature, 2005, 436, 1139 CrossRef CAS PubMed; (b) C. Müller, A. Bauer and T. Bach, Angew. Chem., Int. Ed., 2009, 48, 6640 CrossRef PubMed; (c) C. Müller, A. Bauer, M. Maturi, M. C. Cuquerella, M. A. Miranda and T. Bach, J. Am. Chem. Soc., 2011, 133, 16689 CrossRef PubMed.
- For selected examples, see ref. 2 and 5 and (a) J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160 CrossRef CAS PubMed; (b) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875 CrossRef CAS PubMed; (c) M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem.–Eur. J., 2012, 18, 7336 CrossRef CAS PubMed.
- For recent reviews, see: (a) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534 CrossRef CAS PubMed; (b) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed.
- For a recent review, see: A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC.
- Visible light photoredox catalysis has been successfully combined with other means of catalysis. For examples with transition metals, see: (a) D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566 CrossRef CAS PubMed; (b) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034 CrossRef CAS PubMed; (c) M. Rueping, R. M. Koenigs, K. Poscharny, D. C. Fabry, D. Leonori and C. Vila, Chem.–Eur. J., 2012, 18, 5170 CrossRef CAS PubMed; (d) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505. CrossRef PubMed For an example with enamine catalysis, see: M. Rueping, C. Vila, R. M. Koenigs, K. Poscharny and D. C. Fabry, Chem. Commun., 2011, 47, 2360. For examples with Brønsted acid catalysis, see: (a) L. Ruiz Espelt, E. M. Wiensch and T. P. Yoon, J. Org. Chem., 2013, 78, 4107; (b) M. Neumann and K. Zeitler, Chem.–Eur. J., 2013, 19, 6950.
- For a recent review on tertiary amine oxidation using photoredox catalysis, see: L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC.
- For reviews on β-amino acids, see: (a) D. Seebach, D. F. Hook and A. Glättli, Biopolymers, 2006, 84, 23 CrossRef CAS PubMed; (b) R. P. Cheng, S. H. Gellman and W. F. de Grado, Chem. Rev., 2001, 101, 3219 CrossRef CAS PubMed; (c) D. Seebach, T. Kimmerlin, R. Sebesta, M. A. Campo and A. K. Beck, Tetrahedron, 2004, 60, 7455 CrossRef CAS PubMed; (d) D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366 CrossRef CAS PubMed; (e) B. Weiner, W. Szymanski, D. B. Janssen, A. J. Minnaarda and B. L. Feringa, Chem. Soc. Rev., 2010, 39, 1656 RSC; (f) Enantioselective Synthesis of β-Amino Acids, ed. E. Juaristi and V. A. Soloshonok, Wiley, Hoboken, NJ, 2005 Search PubMed.
- For selected examples of biologically active tetrahydroisoquinolines, see: (a) J. R. F. Allen and B. R. Holmstedt, Phytochemistry, 1980, 19, 1573 CrossRef CAS; (b) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS PubMed; (c) V. G. Kartsev, Med. Chem. Res., 2004, 13, 325 CrossRef CAS; (d) J. Renaud, S. F. Bischoff, T. Buhl, P. Floersheim, B. Fournier, M. Geiser, C. Halleux, J. Kallen, H. Keller and P. Ramage, J. Med. Chem., 2005, 48, 364 CrossRef CAS PubMed; (e) K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444 RSC; (f) M. Ludwig, C. E. Hoesl, G. Höfner and K. T. Wanner, Eur. J. Med. Chem., 2006, 41, 1003 CrossRef CAS PubMed; (g) L. Grycová, J. Dostál and R. Marek, Phytochemistry, 2007, 68, 150 CrossRef PubMed.
- D. B. Freeman, L. Furst, A. G. Condie and C. R. J. Stephenson, Org. Lett., 2012, 14, 94 CrossRef CAS PubMed.
- For selected examples of amine oxidation using photoredox catalysis, see: (a) A. G. Condie, J. C. Gonzalez-Gomez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464 CrossRef CAS PubMed; (b) M. Rueping, S. Zhu and R. M. Koenigs, Chem. Commun., 2011, 47, 8679 RSC; (c) D. P. Hari and B. König, Org. Lett., 2011, 13, 3852 CrossRef CAS PubMed; (d) Y.-Q. Zhou, L.-Q. Lu, L. Fu, N.-J. Chang, J. Rong, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2011, 50, 7171 CrossRef PubMed; (e) P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672 CrossRef CAS PubMed; (f) C. Dai, F. Meschini, J. M. R. Narayanam and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 4425 CrossRef CAS PubMed; (g) S. Zhu, A. Das, L. Bui, H. Zhou, D. P. Curran and M. Rueping, J. Am. Chem. Soc., 2013, 135, 1823 CrossRef CAS PubMed.
- For other studies of tetrahydroisoquinoline derivatives in direct C–H oxidation reactions, see: (a) S.-I. Murahashi, N. Komiya, H. Terai and T. Nakae, J. Am. Chem. Soc., 2003, 125, 15312 CrossRef CAS PubMed; (b) M. North, Angew. Chem., Int. Ed., 2004, 43, 4126 CrossRef CAS PubMed; (c) Z. Li and C.-J. Li, Eur. J. Org. Chem., 2005, 3173 CrossRef CAS; (d) S.-I. Murahashi, N. Komiya and H. Terai, Angew. Chem., Int. Ed., 2005, 117, 7091 CrossRef; (e) Z. Li and C.-J. Li, J. Am. Chem. Soc., 2005, 127, 3672 CrossRef CAS PubMed; (f) Z. Li and C.-J. Li, J. Am. Chem. Soc., 2005, 127, 6968 CrossRef CAS PubMed; (g) A. J. Catino, J. M. Nichols, B. J. Nettles and M. P. Doyle, J. Am. Chem. Soc., 2006, 128, 5648 CrossRef CAS PubMed; (h) Z. Li, D. S. Bohle and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8928 CrossRef CAS PubMed; (i) K. M. Jones and M. Klussmann, Synlett, 2012, 23, 159 CAS.
- See the ESI† for catalysts evaluated in addition to the catalysts presented in Table 1.
- (a) S. E. Reisman, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 7198 CrossRef CAS PubMed; (b) R. R. Knowles, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 5030 CrossRef CAS PubMed; (c) J. A. Birrell, J.-N. Desrosiers and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 13872 CrossRef CAS PubMed; (d) S. Lin and E. N. Jacobsen, Nat. Chem., 2012, 4, 817 CrossRef CAS PubMed.
- For addition of silyl ketene acetals to N-acylquinolinium ions, see: M. S. Taylor, N. Tokunaga and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 6700 CrossRef CAS PubMed . The class of catalysts providing the optimal enantioselection for these substrates were ineffective in the present study.
- Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187 RSC.
- There is a single report describing the oxidative deprotection of the product of methyllithium addition to an N-PMP protected tetrahydroisoquinoline, see: D. Taniyama, M. Hasegawa and K. Tomioka, Tetrahedron: Asymmetry, 1999, 10, 221 CrossRef CAS.
- V. Jurčík, K. Arai, M. M. Salter, Y. Yamashita and S. Kobayashi, Adv. Synth. Catal., 2008, 350, 647 Search PubMed.
- (a) C. L. Wong and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 5593 CrossRef CAS; (b) K. L. Rollick and J. K. Kochi, J. Am. Chem. Soc., 1982, 104, 1319 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectroscopic data of all new compounds. See DOI: 10.1039/c3sc52265b |
| This journal is © The Royal Society of Chemistry 2014 |