 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
3D optical imaging of multiple SERS nanotags in cells†
Sarah
McAughtrie‡
a,
Katherine
Lau‡
b,
Karen
Faulds
a and
Duncan
Graham
*a
aCentre for Molecular Nanometrology, WestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: duncan.graham @strath.ac.uk; Fax: +44 (0)141 5520876; Tel: +44 (0)141 5484701
bRenishaw plc, Spectroscopy Products Division, Old Town, Wotton-Under-Edge, GL12 7DW, UK
First published on 10th July 2013
Abstract
Recent studies have clearly demonstrated that Raman and surface enhanced Raman scattering (SERS) spectroscopies are information rich, non-destructive techniques for the monitoring of subtle intracellular changes. However, despite the demonstrated and sophisticated applications of these techniques in cell studies there still remains a lack of accompanying 3D images. Herein we demonstrate for the first time combined 3D Raman and SERS imaging for the simultaneous confirmation of the cellular inclusion and multiple component detection of SERS nanotags. We also report on the 3D elucidation of the cell nuclei by multivariate analysis methods. Imaging in 3D will be critical to understanding architectural changes between diseased and healthy cells, and tissues. It will also provide non-destructive definitive proof of cellular uptake whilst simultaneously confirming targeting of SERS nanotags to their intended destinations.
Introduction
The application of Raman and surface enhanced Raman scattering (SERS) spectroscopies to complex biological and medical samples has increased dramatically in recent years. This is due in part to improvements in instrument sensitivity but also because both represent a non-destructive method by which considerable information can be gained. Raman spectroscopy also has the added advantage of being a non-invasive analysis method. Whilst the Raman spectra of biological samples are often complex, subtle changes between samples can be readily elucidated by the incorporation of multivariate analysis methods. For example, Raman spectroscopy has been used to identify cell components1 and to discriminate between cancerous and normal cells,2 cancer cells with differing phenotype3 and distinguishing human embryonic stem cells from differentiated cells.4 In the case of SERS, unfunctionalised nanoparticles have been used to obtain information about cell compartments,5 endocytosis,6 as well as cell health7 and viability.8 Function specific SERS nanotags have also been designed for the molecular profiling of single cells,6 intracellular pH sensing9 and in vivo tumour targeting and detection.10,11Despite the advanced applications of both Raman and SERS spectroscopies there are limited examples of sophisticated 3D imaging within the literature to accompany these applications.12 Imaging in 3D by Raman spectroscopy can be relatively simple to achieve using depth profiling in the z direction and software packages such as ImageJ (National Institutes of Health (NIH)) and Volocity™ (Perkin Elmer®). 3D imaging has the potential to provide a wealth of information which could be critical to the detection of disease states. For example, 3D Raman imaging of cells and tissues which results in the resolution of cell organelles represents a non-invasive technique for studying cell construction12 and this could be extremely beneficial when investigating changes in cellular architecture between diseased and healthy cells. If 3D Raman were combined with 3D SERS imaging it may be possible to track nanotags functionalised with specific organelle targeting moieties or drug molecules. Imaging in this manner would not only demonstrate non-destructive evidence of cellular uptake but it would provide evidence that the tags are being directed to their intended targets.
Indeed one of the most notable difficulties when using SERS nanotags for intracellular imaging is in determining cellular inclusion. Whilst it is straightforward to track the ‘uptake’ and localisation of the nanotags by SERS mapping in two dimensions,13,14 it provides no real evidence of uptake and thus cellular inclusion. Traditionally, internalisation has been confirmed by transmission electron microscopy (TEM) or depth-profiling in the z-direction. However, TEM imaging requires convoluted sample preparation, the images are often expensive to obtain and therefore the approach is not open to all. In addition, obtaining a SERS map of the same cell examined by TEM is very challenging and results in the destruction of the cell rendering it unsuitable for live cell analysis. Whilst conventional TEM imaging can provide evidence of cellular inclusion confocal scanning transmission electron microscopy (STEM)15 as well as X-ray diffraction16 and topography,17 and ion-abrasion scanning electron microscopy (SEM)18 can be used to generate 3D cell images. Although these techniques provide excellent resolution (nm range cf. μm range for Raman spectroscopy), comparable acquisition times (hours) and the deduction of cell organelles in 3 dimensions no biochemical information is gained and some of these techniques can result in cellular destruction. The generation of the 3D images also relies on the post collection processing of stacked 2D images. With regards to fluorescence microscopy for the generation of 3D cell images resolution has been greatly enhanced (∼30 nm in the x, y and z directions cf. μm)19 with the advent of approaches such as stimulated emission depletion (STED) microscopy19 and structured illumination microscopy (SIM).20 Acquisition times are considerable faster since the collection of a full vibrational spectrum is not required, however, as with the other techniques fluorescence microscopy is limited in that it prohibits biochemical imaging of the cell contents and the 3D images are again generated from 2D stacks. In addition fluorophores, unlike Raman active labels, can be prone to photobleaching which is problematic if they are to be used for 3D imaging where the acquisition of successive z-sections is necessary and where any bleaching could compromise the correct reconstruction of 3D structures.21 As fluorophores exhibit broad absorption and emission bands the detection of multiple components within a sample can be compromised. This is not an issue when employing Raman or SERS active labels since sharp (nm width)22 molecularly specific bands are produced. Furthermore, the use of SERS active labels can confer picomolar sensitivity in vivo as opposed to nanomolar sensitivity when quantum dots and conventional fluorescence microscopy are employed.23 Here, we demonstrate for the first time the concept of 3D Raman and SERS imaging for the simultaneous confirmation of cellular inclusion and multiple component detection, thus avoiding the need for expensive TEM images. Novelty therefore arises from the fact that specific molecular information can be garnered and from the fact, unlike many of the techniques mentioned, the entire volume is collected at once, all the collected data can be processed and analysed as one data set, and both uni-variate and multivariate analyses can be applied to the volume. The advantage of applying statistical evaluation to the entire volume is that the trends in the data are explained in the x, y and z directions simultaneously, as opposed to applying multivariate analysis to each plane and reassembling plane images as a block which takes only trends in the x and y directions into account.12
Results and discussion
A multiple component suspension of four nanotags24 (nanotag characterisation data – Fig. S1 and 2 and Table S1 and 2†) (4-mercaptopyridine (MPY), 5′5-dithiobis(2-nitrobenzoic acid) (DTNB), 4-nitrobenzenethiol (NBT) and 2 naphthalenethiol (2-NPT) labelled nanotags – ESI†) was applied to Chinese Hamster Ovarian (CHO) cells according to the standard protocol (ESI†). Cell samples were then mapped in 2D (StreamLine™, StreamLineHR™ – Renishaw inVia Raman spectrometer/Leica DM 2500 microscope, Renishaw plc, Gloucestershire, UK.) to rapidly identify cells which had taken up the suspension and in which all four or multiple components could be identified (data not shown). The nanotags were selected on the basis that aggregated systems formed following the addition of 1,6-hexamethylenediamine (HMD) to the silver citrate nanoparticles.24 Small clusters such as this are ideal scaffolds for the formation of hotspots24,25 and thus enhanced SERS.25 Previous investigations (data not shown) have also shown that signals can decrease 10–100 fold following cell uptake so an aggregated system is necessary if the nanotags are to be detected intracellularly. However, in order to ensure that aggregation is slowed and to retard the formation of excessively large clusters which would not be suitable for cellular uptake26,27 polyvinylpyrrolidone (PVP) was added to quench the aggregation.24 The polymer layer stabilises the hot spots24 whilst still allowing the small molecule reporters to infiltrate these zones.24 The small molecule reporters (MPY, DTNB, NBT and 2-NPT) were selected on the basis that no synthesis was required, they like the other nanotag materials, are inexpensive, are readily available, give strong SERS signals, nanoparticle conjugation – via terminal thiol groups or disulfide linkages – is extremely straightforward and their chemical structures are such that subsequent functionalisation with biomolecules for the targeting of specific cell components is relatively straightforward. In addition to this the small molecule reporters9,13,14,24,28 have been widely used in intracellular investigations demonstrating the suitability of the nanotags for cellular uptake. Nanotag identification was a two-fold process whereby each of the components was identified from their unique peaks (Fig. S2†) and direct classical least squares (DCLS) multivariate analysis. This method is typically employed when reference spectra are available for all of the components and involves fitting the unknown data (collected during mapping) to a linear combination of the specified component spectra (standard suspension spectra). If a match was found to occur between the standard and the collected spectra a false colour was assigned and separate false colour images were created for each component. The contributions were normalised to a value of between 0 and 1 where values close to 1 indicate areas within the image with high similarities in shape to that of the reference spectrum. Once a cell containing all four or multiple components had been identified, it was imaged in 3D.To successfully image a multiple component positive cell in 3D and therefore verify the uptake of the nanotags, the cell was mapped using volume mapping, a confocal configuration which allows layers of Raman and SERS images to be rapidly collected. When this method is employed the step sizes in the x and y directions can be defined independently from the step size in the z direction. Here the step sizes in the x and y directions were set at 0.8 μm and 0.5 μm for the cell and nanotag maps respectively, and in the z direction it was set at 1 μm. The z-range was defined as ±3 μm from the point of focus when the cell was observed under white light illumination (set as z = 0). The cell sample was imaged under immersion in a saline solution using an Olympus 60× (N.A. 1) water immersion objective. In order to volume map both the cells and the SERS nanotags two different laser excitations were required – one where the intrinsic Raman signals of the cell dominate and there is minimal interference from the SERS nanotags, and one where the SERS signals from the nanotags are most prevalent. The total acquisition times for the cell (0.8 μm step size, 2 s acquisition) and the nanotag (0.5 μm step size, 0.5 s acquisition) maps were 3.24 h and 4.7 h respectively. This is comparable with other Raman studies12 and unlike the depth profiling methods there is no need for the post collection processing of 2D stacks to generate 3D images.12 Additionally, in comparison to other methods although the overall collection time is somewhat greater this is offset by the biochemically specific information obtained. The 3D volume Raman cell map was generated (Fig. 1d–i) by mapping the highlighted cells (Fig. 1a–c) using 532 nm excitation.
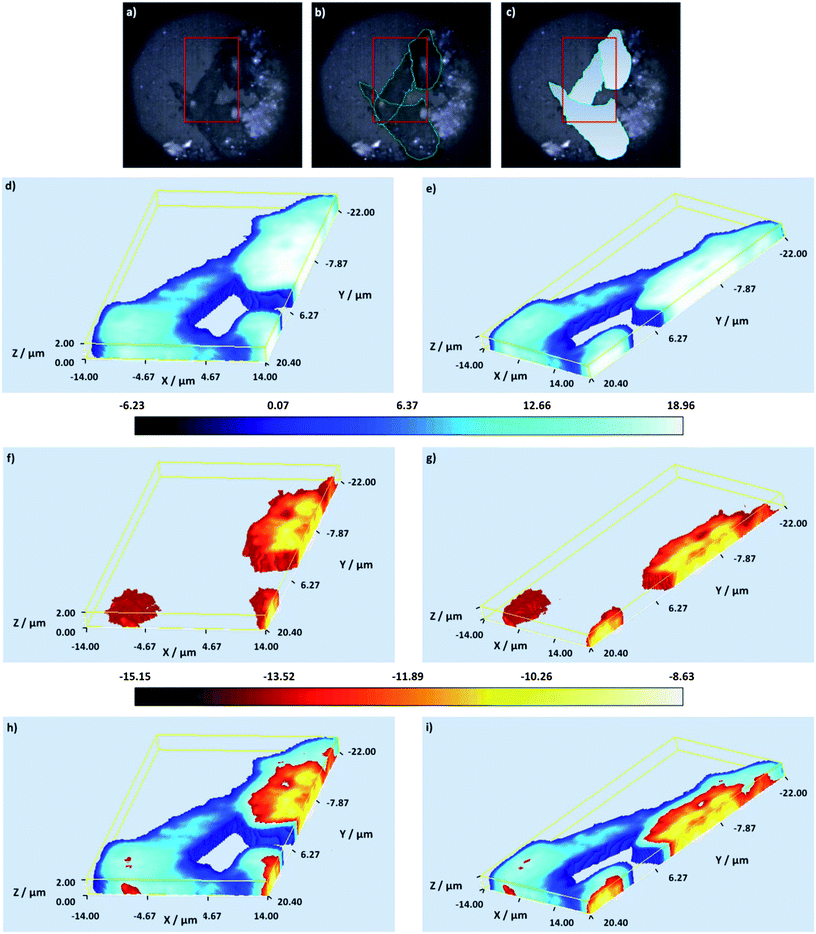 | ||
| Fig. 1 White light and false colour volume 3D Raman images of the mapped cells – (a) white light image of the mapped cells, the cells are outlined in green in (b) and the mapped cell areas are distinguished with a gradient fill in blue in (c). In (a)–(c) the red box indicates the complete 3D Raman analysed area. (d and e) A false colour 3D Raman cell image generated using PC 2 displayed at different angles. (f and g) A false colour 3D Raman nuclei image generated using PC 1 displayed at different angles. (h and i) Combined false colour 3D Raman cell and nuclei images presented at different angles. In (d)–(g) the false colour scale bar is shown below the images. | ||
At this wavelength intrinsic Raman signals from cell components such as proteins, amino acids, lipids and DNA can be readily visualised.5,6 Following data collection, the false colour Raman 3D images were constructed by performing uni-variate analysis based on signal to baseline maps (the integrated intensities of selected bands), and multivariate analysis based on principal component analysis (PCA). A signal to baseline map was generated specifically from the ring breathing mode of the amino acid phenylalanine (998–1008 cm−1, ∼1004 cm−1)5,6 (data not shown) and this was found to clearly define the cell structure.
Phenylalanine is found ubiquitously in the cell hence it is an excellent marker for its delineation. However, as we also wished to determine the location of the SERS nanotags in relation to the cell organelles, PCA was applied to the data (normalised, not mean centred) to determine if there were any spectral variations, which could be ascribed to specific cell organelles.29 The main principles of PCA have been previously described29,30 but for this particular data set the PCA conditions included spectrum centring and normalisation. Mean centring was not applied since the band positions were sufficiently different to distinguish the two PCs chemically (Fig. 2). The main advantages of these conditions are that the loadings reflect the baseline offset, the background and the appearance of a typical spectrum. For the mapped cells two principal components (PCs) explained 86.5% of the variance in the data set with PCs 1 and 2 representing the cell contents (Fig. 2a–d). When comparing the PC loadings (Fig. 2d) and the band assignments (Table 1) PC 2 is characterised collectively by Raman bands attributed to protein, lipid and nucleic acids but most notably by peaks at 1004 cm−1, 1245 cm−1 and 1451 cm−1 assigned to phenylalanine,31,32 amide III (protein) band33 and the CH2CH3 deformation of protein and lipid.34 PC2 therefore represents the general cellular regions. This is consistent with the raw Raman spectra (Fig. 2c) and the PC2 image clearly defines the structure of the mapped cells (Fig. 1d, e, h and i).
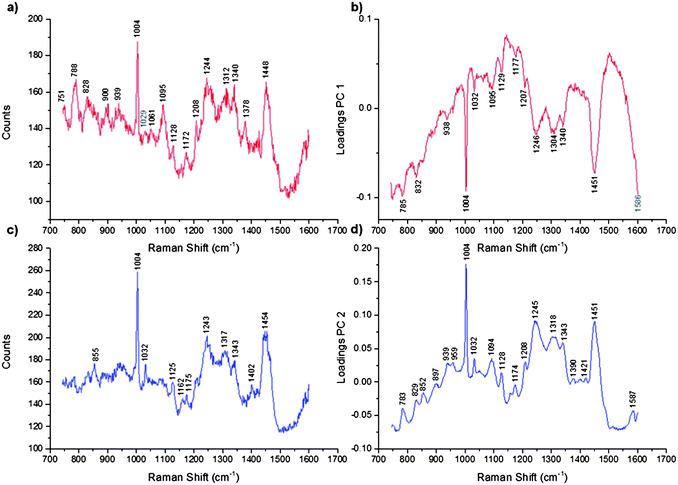 | ||
| Fig. 2 Representative Raman spectra and the corresponding PC loadings for the cells and the nuclei. (a) A representative Raman spectrum measured from the nuclei, (b) PC 1 loadings which distinguish the nuclei, (c) representative Raman spectrum measured from the cells and (d) PC 2 loadings which distinguish the cells. | ||
| Peak position (cm−1) | Assignment |
|---|---|
| 783 | DNA |
| 785 | νO–P–O stretch DNA |
| 829 | O–P–O stretch DNA/RNA ring breathing tyrosine |
| 832 | Tyrosine, νasymO–P–O |
| 852 | Proline/tyrosine |
| 897 | — |
| 938/939 | νC–C, peptide |
| 959 | — |
| 1004 | Phenylalanine |
| 1032 | CH2CH3 collagen and phospholipids, phenylalanine |
| 1094/1095 | DNA, νC–N lipid |
| 1128 | C–N (proteins), C–O (carbohydrates) |
| 1174 | Phenylalanine, tyrosine |
| 1177 | Tyrosine, cytosine, guanine |
| 1207/1208 | Phenylalanine |
| 1245/1246 | Amide III |
| 1304 | CH2 deformation (lipid) adenine/cytosine |
| 1318 | Guanine (DNA/RNA) |
| 1340 | — |
| 1421 | Adenine, guanine ring breathing modes of DNA/RNA |
| 1451 | CH2CH3 deformation |
| 1586/1587 | Phenylalanine |
The PC1 domain contains negative scores and is thus characterised by the negative peaks in the loadings. PC1 is characterised by a combination of peaks ascribed to nucleic acids, nucleotides, amino acids and protein. These include phenylalanine peaks31,32 at 1004 cm−1 and the CH2CH3 deformation of protein at 1451 cm−1 (Fig. 2b).34 The PC loadings suggest that PC1 represents domains which are biochemically distinct from the main cell structure and based on these loadings we propose that PC1 represents the cell nuclei (Fig. 1f–i). This is consistent with the raw Raman spectra from the nuclei, specifically DNA peaks at 751 cm−1 and 788 cm−1 (Fig. 2a).30,35
The 3D volume SERS nanotag map was generated by mapping the same volume as highlighted (Fig. 1b and c) but with 633 nm excitation instead. However, because of the smaller step size used (0.5 μm cf. 0.8 μm) the SERS image was collected at 0.5 μm higher in the y direction than the Raman cell image. This has been corrected for in the final 2D images (ESI†). At this wavelength the signals from the small molecule reporters can be readily visualised and using the employed laser power intrinsic Raman signals from the cells is minimal. Following data collection the false colour SERS 3D images were constructed by performing multivariate data analysis in the form of component DCLS.
The two independently collected 3D data sets (cells and nanotags) were then combined in 2 dimensions for the determination of cellular uptake and multiple component detection (Fig. 3). The z range was defined as ±3 μm from the point of focus for both the cell and the nanotag maps. However, when the volume 3D cell map was collected (data not shown) it was found that only certain z-slices were of relevance. This was ascertained by viewing the 3D volume cell map along the z-direction. From this angle it was apparent that the spectra from above z-slice +2.00 and below z-slice +0.00 were not of relevance. In light of this the 3D cell map was recollected from z-slice +2.00 to +0.00 but the 3D nanotag map was collected from the entire z-range previously mentioned. Despite this it was still possible to combine the relevant z-slices in 2D to ascertain cellular uptake, and the combined cell and nanotag z-slices are presented in Fig. 3d–f.
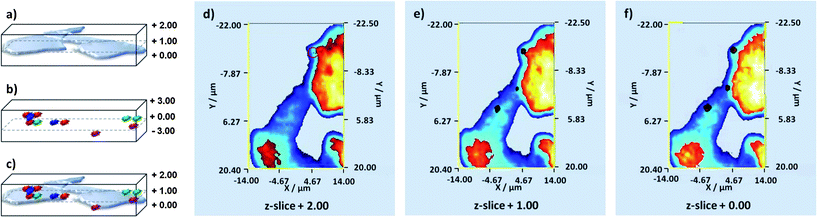 | ||
| Fig. 3 Schematic of how the two volume 3D data sets are combined. (a) Schematic of volume 3D Raman cell map, (b) schematic of volume 3D SERS nanotag map, (c) combined 3D volume Raman cell and SERS nanotag map. Images from each plane of the 3D volume Raman cell and SERS nanotag maps can be extracted and these can be combined in 2D – (d) z-slice nanotags and cell +2.00, in this z-slice the signal is considerably weaker than those obtained from the other z-slices and as such the corresponding false colour is considerably smaller hence it has been highlighted with an appropriately coloured circle, (e) z-slice nanotags and cell +1.00 and (f) z-slice nanotags and cell +0.00. MPY, DTNB and NBT labelled nanotags are shown in blue, green and red respectively. | ||
On close inspection of the combined z-slices we can instantly determine that three out of the four nanotags, MPY, DTNB and NBT labelled nanotags, which are shown in blue, green and red respectively, within the multiple component suspension are located within the cells (Fig. 3d–f). In z-slice +2.00 only NBT labelled nanotags were observed (Fig. 3d), whilst in z-slice +1.00 only NBT and DTNB labelled nanotags were found (Fig. 3e) and in z-slice +0.00 three out of the four nanotags, MPY, DTNB and NBT, were observed (Fig. 3f). The reasons as to why 2-NPT labelled nanotags were not observed intracellularly are unclear but if 2-NPT remains extracellularly the possibility of using this nanotag as an external standard exists. On the basis of these images (Fig. 3d–f) it is quite difficult to observe the three nanotags located within the cells because there is a degree of co-localisation. In theory, co-localisation may be expected since the suspension applied to the cell population contained equal proportions of the four individually labelled tags which were well mixed. Thus there was an equal probability of each nanotag within the suspension being internalised simultaneously. As such z-slice +0.00 has been enlarged (Fig. 4a–c) for ease of observation and the combined SERS z-slice image for the three nanotags and for each of the individually labelled nanotags located intracellularly are also shown (Fig. 4d).
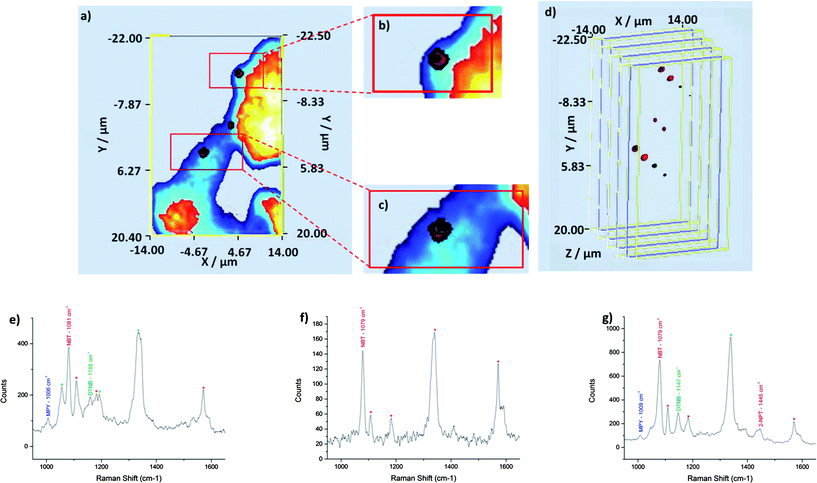 | ||
| Fig. 4 Enlarged image of z-slice +0.00 and the corresponding SERS spectra for each of the three false colour clusters. (a) 2D z-slice nanotags and cell +0.00, (b) and (c) enlarged areas from the top and bottom clusters respectively, z-slice +0.00 which shows three of the four nanotags within a single area and a single cell and (d) exploded 3D SERS z-slice image for the three nanotags combined and for each of the individual nanotags. In the enlarged image it is difficult to identify the three false colours because they overlap but in the exploded image each can be clearly visualised. (e–g) Representative SERS spectra obtained for the top, middle and bottom false colour clusters. | ||
Each of the components was identified by their unique peak and component DCLS analysis. Unique peaks were found at ∼1004 cm−1, 1150 cm−1, 1084 cm−1 and 1450 cm−1 for MPY, DTNB, NBT and 2-NPT respectively (Fig. S2†). With the component DCLS a ‘false’ colour was assigned if a match was found to occur between the reference and the collected data. In order to further confirm the spatial positioning of the components, the data was viewed on moveable plane slices within the volume. When viewed in this way the corresponding spectra for a selected position are also displayed and these can be extracted and visually compared with standard spectra from the nanotag suspensions.
For z-slice +0.00 representative SERS spectra have been extracted for each of the three locations where the nanotags were found (Fig. 4e–g). In accordance with the co-localisation observed, cumulative SERS signals concomitantly arise from the nanotags. That is, we observe SERS signals from all of the nanotags which are found within a specified location (Fig. 4e–g). The unique peak for the appropriate nanotag has been labelled with the corresponding abbreviation and peak position. The minor peaks have also been tentatively assigned to a small molecule reporter and labelled with an asterisk of corresponding colour (Fig. 4e–g). For the top spot in z-slice +0.00 MPY, NBT and DTNB were identified at ∼1006 cm−1, ∼1081 cm−1 and ∼1158 cm−1 respectively. For the middle spot, NBT was identified at ∼1079 cm−1. MPY, NBT and DTNB were identified at ∼1008 cm−1, ∼1079 cm−1, ∼1147 cm−1 in the bottom cluster. 2-NPT was also identified at ∼1445 cm−1 in the bottom spot although no false colour has been assigned to 2-NPT at that particular location.
It should also be noted that none of the nanotags penetrated the nuclei (Fig. 3 and 4) and they presumably resided in the cytoplasm or endosome compartments. This is to be expected since the nanotags were not functionalised with targeting moieties specific for this or any other cell organelle. Based on this information these unfunctionalised nanotags could be used as controls to distinguish between targeting and non-targeting nanotags when targeting of the nucleus is attempted. This will be investigated further in later studies.
Conclusions
In conclusion, volume 3D Raman and SERS imaging represents a non-destructive technique for the simultaneous confirmation of cellular inclusion and multiple component detection. Three of the four components of the suspension were detected, and by extracting and combining 2D z-slices from the 3D images it was possible to determine the intracellular location of the nanotags without the need for expensive and destructive TEM imaging. This study is also the first to report on the generation of true 3D Raman and 3D SERS images. Although, other studies have reported the production of 3D images12 these have been constructed using depth profiling methods. In the current study the entire volume was analysed at once reflecting the novelty of the technique. It was also possible to define a subcellular compartment without the need for any special sample preparation (i.e. staining). The resolution of further cell organelles12 is expected and the ability to detect such structures whilst maintaining the integrity of the cell will undoubtedly be of benefit to the detection of disease process in which the architecture of the cell changes considerably. The adoption of a multi-marker approach is also critical if disease states are to be fully characterised or multiple cell organelles are to be targeted. Hence, the ability to detect these multi-markers in 3 dimensions is of huge importance for a whole host of applications including cellular disease or tumour detection and the delivery of drugs and their subsequent fate within the cells. This work also has implications for the application of combined 3D Raman and SERS imaging to living cells.Acknowledgements
SMcA acknowledges Renishaw plc for funding. DG acknowledges support from Royal Society Wolfson Trust Research Merit Award.References
- C. Krafft, T. Knetschke, A. Siegner, R. H. W. Funk and R. Salzer, Vib. Spectrosc., 2003, 32, 75–83 CrossRef CAS.
- M. S. Bergholt, W. Zheng, K. Lin, K. Y. Ho, M. Teh, K. G. Yeoh, J. B. Yan So and Z. Huang, Int. J. Cancer, 2011, 128, 2673–2680 CrossRef CAS.
- Y. Oshima, H. Shinzawa, T. Takenaka, C. Furihata and H. Sato, J. Biomed. Opt., 2010, 15, 017009 CrossRef.
- F. C. Pascut, H. T. Goh, N. Welch, L. D. Buttery, C. Denning and I. Notingher, Biophys. J., 2011, 100, 251–259 CrossRef CAS.
- K. Kneipp, A. S. Haka, H. Kneipp, K. Badizadegan, N. Yoshizawa, C. Boone, K. E. Shafer-Peltier, J. T. Motz, R. R. Dasari and M. S. Feld, Appl. Spectrosc., 2002, 56, 150–154 CrossRef CAS.
- J. Kneipp, H. Kneipp, M. McLaughlin, D. Brown and K. Kneipp, Nano Lett., 2006, 6, 2225–2231 CrossRef CAS.
- S. C. Pînzaru, L. M. Andronie, I. Domsa, O. Cozar and S. Astilean, J. Raman Spectrosc., 2008, 39, 331–334 CrossRef.
- H.-W. Tang, X. B. Yang, J. Kirkham and D. A. Smith, Appl. Spectrosc., 2008, 62, 1060–1069 CrossRef CAS.
- A. Pallaoro, G. B. Bruan, N. O. Reich and M. Moskovits, Small, 2010, 6, 618–622 CrossRef CAS.
- X. Qian, X.-H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS.
- C. L. Zavaleta, B. R. Smith, I. Walton, W. Doering, G. Davis, B. Shojaei, M. J. Natan and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13511–13516 CrossRef CAS.
- K. Majzner, A. Kaczor, N. Kachamakova-Trojanowska, A. Fedorowicz, S. Chlopicki and M. Baranska, Analyst, 2013, 138, 603–610 RSC.
- M. K. Gregas, J. P. Scaffidi, B. Lauly and T. Vo-Dinh, Appl. Spectrosc., 2010, 64, 858–866 CrossRef CAS.
- M. K. Gregas, F. Yan, J. Scaffidi, H.-N. Wang and T. Vo-Dinh, Nanomedicine, 2011, 7, 115–122 CrossRef CAS.
- J. J. Einspahr and P. M. Voyles, Ultramicroscopy, 2006, 106, 1041–1052 CrossRef CAS.
- H. Jiang, C. Song, C.-C. Chen, R. Xu, K. S. Raines, B. P. Fahimian, C.-H. Lu, T.-K. Lee, A. Nakashima, J. Urano, T. Ishikawa, F. Tamanoi and J. Miao, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11234–11239 CrossRef CAS.
- W. Gu, L. D. Etkin, M. A. Le Gros and C. A. Larabell, Differentiation, 2007, 75, 529–535 CrossRef CAS.
- J. A. W. Heymann, D. Shi, S. Kim, D. Bliss, J. L. S. Milne and S. Subramaniam, J. Struct. Biol., 2009, 166, 1–7 CrossRef CAS.
- R. Schmidt, C. A. Wurm, A. Punge, A. Egner, S. Jakobs and S. W. Hell, Nano Lett., 2009, 9, 2508–2510 CrossRef CAS.
- L. Schermelleh, P. M. Carlton, S. Haase, L. Shao, L. Winoto, P. Kner, B. Burke, M. C. Cardoso, D. A. Agard, M. G. L. Gustafsson, H. Leonhardt and J. W. Sedat, Science, 2008, 320, 1332–1336 CrossRef CAS.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS.
- J. A. Dougan and K. Faulds, Analyst, 2012, 137, 545–554 RSC.
- S. Keren, C. Zavaleta, Z. Cheng, A. de la Zerda, O. Gheysens and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5844–5849 CrossRef CAS.
- G. B. Braun, S. J. Lee, T. Laurence, N. Fera, L. Fabris, G. C. Bazan, M. Moskovits and N. O. Reich, J. Phys. Chem. C, 2009, 113, 13622–13629 CAS.
- R. Jin, Angew. Chem., Int. Ed., 2010, 49, 2826–2829 CrossRef CAS.
- F. Zhao, Y. Zhao, Y. Liu, X. Chang, C. Chen and Y. Zhao, Small, 2011, 7, 1322–1337 CrossRef CAS.
- L. Y. T. Chou, K. Ming and W. C. W. Chan, Chem. Soc. Rev., 2011, 40, 233–245 RSC.
- N. M. S. Sirimuthu, C. D. Syme and J. M. Cooper, Chem. Commun., 2011, 47, 4099–4101 RSC.
- K. Lau, A. Hobro, T. Smith, T. Thurston and B. Lendl, Vib. Spectrosc., 2012, 60, 34–42 CrossRef CAS.
- D.-H. Kim, R. M. Jarvis, Y. Xu, A. W. Oliver, J. W. Allwood, L. Hampson, I. N. Hampson and R. Goodacre, Analyst, 2010, 135, 1235–1244 RSC.
- W.-T. Cheng, M.-T. Liu, H.-N. Liu and S.-Y. Lin, Microsc. Res. Tech., 2005, 68, 75–79 CrossRef CAS.
- R. Malini, K. Venkatakrishma and J. Kurien, Biopolymers, 2006, 81, 179–193 CrossRef CAS.
- G. Shetty, C. Kedall, N. Shepherd, N. Stone and H. Barr, Br. J. Cancer, 2006, 94, 1460–1464 CrossRef CAS.
- R. J. Lakshimi, V. B. Kartha, C. M. Krishna, J. G. R. Solomon, G. Ullas and P. Uma Devi, Radiat. Res., 2002, 157, 175 CrossRef.
- I. Notingher, C. Green and C. Dyer, J. R. Soc., Interface, 2004, 1, 79–90 CrossRef CAS.
- Z. Huang, A. McWilliams, M. Lui, D. I. McLean, S. Lam and H. Zeng, Int. J. Cancer, 2003, 107, 1047–1052 CrossRef CAS.
- N. Stone, C. Kendell, N. Shepherd, P. Crow and H. Barr, Faraday Discuss., 2004, 126, 141–157 RSC.
- A. J. Ruiz-Chica, M. A. Medina, F. Sanchez-Jimenez and F. J. Ramirez, J. Raman Spectrosc., 2004, 35, 93–100 CrossRef CAS.
- J. W. Chan, D. S. Taylor, T. Zwerdling, S. M. Lane, K. Ihara and T. Huser, Biophys. J., 2006, 90, 648–656 CrossRef CAS.
- C. J. Frank, R. L. McCreecy and D. C. B. Redd, Anal. Chem., 1995, 67, 777–783 CrossRef CAS.
- J. Binoy, J. P. Abraham, I. H. Joe, V. S. Jayakumar, G. R. Pettit and O. F. Nielsen, J. Raman Spectrosc., 2004, 35, 939–946 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full preparation details for the nanoparticles, nanotags and cells are included. The procedures for analysing the cell samples and characterisation data for the nanotags are also included. See DOI: 10.1039/c3sc51437d |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2013 |