 Open Access Article
Open Access ArticleA novel approach to the site-selective dual labelling of a protein via chemoselective cysteine modification†
Ramiz I.
Nathani
,
Paul
Moody
,
Vijay
Chudasama
,
Mark E. B.
Smith
,
Richard J.
Fitzmaurice
and
Stephen
Caddick
*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H OAJ, UK. E-mail: VPEnterprise@ucl.ac.uk; Fax: +44 (0)20 7679 7463; Tel: +44 (0)20 3108 5071
First published on 18th June 2013
Abstract
Local protein microenvironment is used to control the outcome of reaction between cysteine residues and 2,5-dibromohexanediamide. The differential reactivity is exploited to introduce two orthogonal reactive handles onto the surface of a double cysteine mutant of superfolder green fluorescent protein in a regioselective manner. Subsequent elaboration with commonly used thiol and alkyne containing reagents affects site-selective protein dual labelling.
The development of accessible chemical methodologies that enable the site-selective labelling of proteins has transformed the study and utility of these complex biomolecules.1 For example, Nε-acetyl-lysine mimetics can be readily introduced in a defined manner to allow researchers to further investigate this key post–translational modification.2 Whereas, in the arena of protein therapeutics, the ability to conjugate a drug, probe or lifetime extension technology to a protein in a controlled manner generates homogeneous biologics with a range of functions.3 The site-selective labelling of proteins is typically achieved by exploiting the unique reactivity of a specific functional group in the protein of interest, e.g. thiols,4 alkenes,5 azides6 or alkynes.7
Even more challenging than the site-selective generation of singly modified proteins is the homogenous modification of a protein in multiple distinct positions with different probes. Access to site-selectively dual labelled proteins offers opportunities to perform a range of structural studies, employing techniques such as Förster resonance energy transfer to understand protein structure.7 Such a methodology would also allow the construction of a range of biologics with dual function, e.g. theranostics,8 or facilitate the optimal conjugation of both a drug, or imaging probe, with a lifetime extension technology.3a
In order to successfully affect site-selective dual labelling of a protein, two chemoselective, biocompatible processes must be carried out using either native functional groups or those which can be readily introduced in a controlled manner. A number of strategies have been developed to address this challenge. However, many are restricted to modification of protein termini, via introduction of an N-terminal cysteine and a C-terminal intein,9 or sortase labelling,10 which can limit their utility.11 Methods which allow for the selective dual modification of non-terminal positions are highly desirable as they offer choice as to the disposition of the modifications across the protein surface, however, this creates additional complexity. In cases where the protein can be expressed as two separate soluble fragments, it is possible to label each fragment individually and then carry out a ligation to generate the desired dual labelled protein.12 Approaches using a cysteine/tetracysteine orthogonal reaction pair have also been employed.13 The selective introduction and modification of two non-natural amino acids provides exquisite selectivities.14 However, such strategies require custom expression strains and tRNAs to generate the protein, which can be time consuming and expensive, and can ultimately result in low expression yields.15 Alternatively, it might be envisaged that, it should be possible to exploit the differential nucleophilicity of thiols in double cysteine mutant proteins. However, this has brought limited success to date due to the heterogeneity of the modified protein.16
In this report we describe a fundamentally novel approach to the site-selective dual labelling of a protein at non-terminal amino acid positions. Our approach is based on the positioning of two cysteine mutants within a protein sequence such that the two cysteines are cleanly converted into two distinct products upon treatment with a single chemical reagent. More specifically, in the work described herein, a double cysteine mutant of a protein is initially transformed into a bis-sulfonium using a simple small molecule reagent. The fate of each sulfonium thereafter is controlled by the protein microenvironment, i.e. the sulfonium can either persist as a stable entity or eliminate to yield dehydroalanine, thus resulting in site-selective dual functionalisation of a protein based on substrate control. As a proof of concept, we have applied this rationale to generate a site-selectively dual labelled superfolder green fluorescent protein.
Bioconjugation via the selective modification of free cysteine in proteins has received a great deal of attention due to the unique reactivity of its thiol side chain,17 and the ease with which the residue can be selectively introduced via site-directed mutagenesis. For example, Davis has described the modification of cysteine 1 to generate dehydroalanine 2, which can then be used for bioconjugation in a range of protein substrates, including a single cysteine mutant (S156C) of the protease subtilisin.18 The formation of dehydroalanine 2 is a consequence of β-elimination of sulfonium 3, generated from reaction of cysteine thiol 1 with 2,5-dibromohexanediamide 4 at pH 8 (Scheme 1).18c However, we have recently discovered that sulfonium 3, derived from a single cysteine mutant of superfolder green fluorescent protein (S147C), can be isolated and subsequently used in an alternate bioconjugation strategy, via a ring opening reaction with azide, to afford an azide functionalised protein 5 (Scheme 1, see Fig. S7 and S8 in the ESI†).19 We rationalised that the ability to control the stability of a protein sulfonium, using the protein's microenvironment, could therefore offer a new approach to site-selective protein dual labelling.
Our study began with an attempt to understand the structural factors effecting the formation of dehydroalanine 2 from a cysteine derived sulfonium 3 based on our previous observations and those reported by Davis.5,18a–c As elimination to dehydroalanine 2 inherently requires loss of the α-proton of sulfonium 3 we envisaged that its microenvironment may play an important role in the fate of the sulfonium species. Interestingly, examination of the crystal structures of subtilisin (PDB ID: 1GCI,20 see ESI†) and superfolder GFP (PDB ID: 2B3P,21Fig. 1) did indeed highlight differences in the environments of the α-protons of the residues S156 and S147, respectively. In GFP the structured protein β-barrel appears to shield the α-proton of S147 (green) rendering it inaccessible, solvent accessible surface area = 0.0 Å2 (calculated using Naccess),21 thus preventing elimination to dehydroalanine 2. However, in subtilisin the S156 α-proton appears to be solvent accessible (see ESI†), solvent accessible surface area = 4.5 Å2,22 and therefore a single cysteine mutant at this position is prone to elimination to give dehydroalanine 2 (Scheme 1).
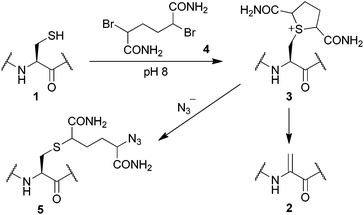 | ||
| Scheme 1 Reaction modes of sulfonium 3. | ||
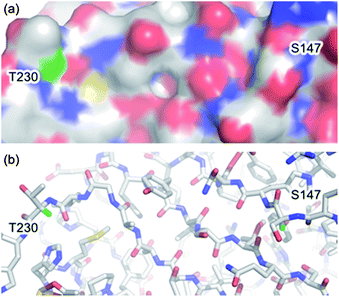 | ||
| Fig. 1 (a) Surface and (b) stick representation of superfolder green fluorescent protein (PDB ID: 2B3P)21 showing α-protons of S147 and T230 (green). Protons were added using PyMOL.23 | ||
Using the same analysis, we identified residue T230 of superfolder GFP (Fig. 1), close to the C-terminus, as having a solvent accessible α-proton (green), solvent accessible surface area = 4.3 Å2.21,22 Thus, in order to evaluate our hypothesis on the significance of α-proton accessibility on the fate of sulfonium, we expressed and treated a single cysteine mutant at position 230, GFP(T230C, 233Δ) 6, with 2,5-dibromohexanediamide (4, 50 eq., 2 h, 37 °C). Gratifyingly, this generated GFP(T230Dha, 233Δ) 7, as determined by LCMS (observed 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 543, expected 28541), cleanly, as a single product after 2.5 h at 37 °C (Scheme 2).†
543, expected 28541), cleanly, as a single product after 2.5 h at 37 °C (Scheme 2).†
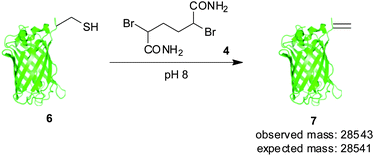 | ||
| Scheme 2 Conversion of GFP(T230C, 233Δ) 6 to GFP(T230Dha, 233Δ) 7. | ||
Equipped with these findings we sought to explore the use of controlled sulfonium elimination as an approach for the site-selective dual labelling of a protein. Thus, we generated double mutant GFP(S147C, T230C, 233Δ) 8. Initially, to explore if there was any difference in the nucleophilicity of the cysteine thiols at positions 147 and 230, the double mutant was treated with a stoichiometric amount of N-methylmaleimide. As a statistical mixture of products was observed (see ESI, Fig. S10†), this confirmed that the cysteine thiols exhibited essentially equivalent nucleophilicity and that any observed chemoselectivity would almost certainly not be due to this factor. Having established this, we proceeded to incubate double mutant GFP(S147C, T230C, 233Δ) 8 with 2,5-dibromohexanediamide (4, 50 eq., 2 h, 37 °C) to see if the results observed on the single mutants at positions 147 and 230 would be translated. To our delight, we observed formation of dual modified GFP(S147Sulf, T230Dha, 233Δ) 9 as the sole identifiable product, as determined by SDS-PAGE and LCMS (observed 28699, expected 28697, Fig. 2). Furthermore, direct treatment of GFP(S147Sulf, T230Dha, 233Δ) 9 with azide (NaN3, >1000 eq., 37 °C, 2 h) resulted in the addition of a single azide to give GFP(S147Azide, T230Dha, 233Δ) 10, a site-selectively dual functionalised protein bearing orthogonal reactive handles, as determined by SDS-PAGE and LCMS (observed 28736, expected 28739, Fig. 2). The regioselectivity of the reaction of GFP(S147Sulf, T230Dha, 233Δ) 9 with azide was ascertained by incubation of a fresh sample of GFP(T230Dha, 233Δ) 7 with sodium azide (>1000 eq., 37 °C, 4 h). Cross reaction of azide with dehydroalanine in 7 was not observed, confirming selective ring opening of position 147 sulfonium in GFP(S147Sulf, T230Dha, 233Δ) 9 generating GFP(S147Azide, T230Dha, 233Δ) 10 (see ESI, Fig. S12†).
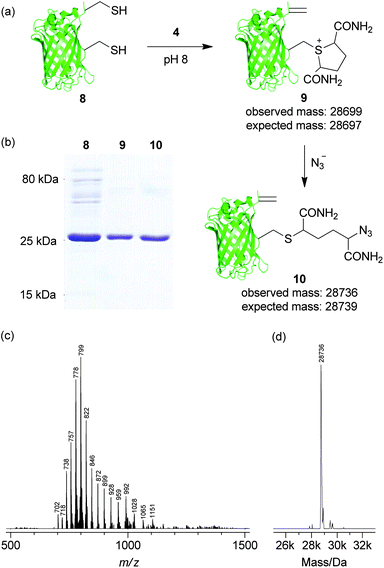 | ||
| Fig. 2 Regioselective dual modification of double cysteine GFP mutant 8. (a) Generation of GFP(S147Azide, T230Dha, 233Δ) 10 from GFP(S147C, T230C, 233Δ) 8. (b) SDS-PAGE characterisation of 8, 9 and 10 with Coomassie staining. (c) Raw and (d) deconvoluted MS data for GFP(S147Azide, T230Dha, 233Δ) 10. | ||
We then sought to demonstrate the utility of azide/dehydroalanine constructs through elaboration of the protein scaffold via reaction with these two orthogonal groups (Scheme 3). Thus, we treated GFP(S147Azide, T230Dha, 233Δ) 10 with commercially available strained alkyne dye, dibenzylcyclooctyne PEG4-Fluor 545 (Jena Biosciences), to affect the formation of the expected dye-GFP conjugate (observed 29679, expected 29675) through a chemoselective strain-promoted alkyne-azide cyloaddition. It has previously been demonstrated that protein dehydroalanines undergo facile conjugation with free thiols, albeit not in a stereodefined manner, allowing introduction of various thiolated probes.19 Thus, subsequent addition of a simple thiol, 2-mercaptoethanol, to the intermediate Dye-GFP conjugate gave dual labelled GFP 11 (observed 29755, expected 29753) decorated with a dye, tetramethylrhodamine, and a thiol in a regioselective manner.†
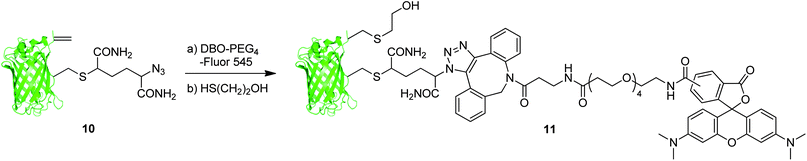 | ||
| Scheme 3 Elaboration of GFP(S147Azide, T230Dha, 233Δ) 10. | ||
Conclusions
A facile and generally accessible methodology for the site-selective dual modification of proteins is a hitherto unmet need and offers a wide range of possibilities for development of new approaches to chemical biology and for therapeutic development. Although other techniques are available to affect the site-selective dual labelling of a protein, they tend to be limited and each suffer from a number of drawbacks. The strategy for chemoselective protein dual labelling described herein enables dual functionalisation using orthogonal reactivity of natural amino acid side chains under mild conditions, underpinned by two sequential chemoselective reactions. We are hopeful that this strategy can be applied to any pair of cysteine residues wherein one residue has an accessible α-proton, and thus readily forms dehydroalanine, and the α-proton of the second residue is sufficiently shielded, and thus persists as a sulfonium that can undergo chemoselective ring opening by the addition of azide. Moreover, the derived azide/dehydroalanine proteins can be further modified in a chemoselective manner using well established methods and commercial reagents to affect site-selective dual labelling in a facile manner.Acknowledgements
The authors gratefully acknowledge the EPSRC, BBSRC, Wellcome Trust, UCL, UCLB and GSK for support of our programme.Notes and references
- (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS; (b) C. T. Walsh, S. Garneau-Tsodikova and G. J. Gatto, Angew. Chem., Int. Ed., 2005, 44, 7342–7372 CrossRef CAS; (c) D. F. Qi, C. M. Tann, D. Haring and M. D. Distefano, Chem. Rev., 2001, 101, 3081–3111 CrossRef CAS; (d) L. M. Artner, L. Merkel, N. Bohlke, F. Beceren-Braun, C. Weise, J. Dernedde, N. Budisa and C. P. R. Hackenberger, Chem. Commun., 2012, 48, 522–524 RSC.
- F. Li, A. Allahverdi, R. Yang, G. B. J. Lua, X. Zhang, Y. Cao, N. Korolev, L. Nordenskiöld and C.-F. Liu, Angew. Chem., Int. Ed., 2011, 50, 9611–9614 CrossRef CAS.
- (a) M. S. S. Palanki, A. Bhat, B. Bolanos, F. Brunel, J. Del Rosario, D. Dettling, M. Horn, R. Lappe, R. Preston, A. Sievers, N. Stankovic, G. Woodnut and G. Chen, Bioorg. Med. Chem. Lett., 2013, 23, 402–406 CrossRef CAS; (b) L. Fass, Mol. Oncol., 2008, 2, 115–152 CrossRef; (c) S. C. Alley, N. M. Okeley and P. D. Senter, Curr. Opin. Chem. Biol., 2010, 14, 529–537 CrossRef CAS.
- (a) D. P. Nguyen, T. Elliott, M. Holt, T. W. Muir and J. W. Chin, J. Am. Chem. Soc., 2011, 133, 11418–11421 CrossRef CAS; (b) P. Moody, M. E. B. Smith, C. P. Ryan, V. Chudasama, J. R. Baker, J. Molloy and S. Caddick, ChemBioChem, 2012, 13, 39–41 CrossRef CAS.
- K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298–304 CrossRef CAS.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS.
- D. P. Nguyen, H. Lusic, H. Neumann, P. B. Kapadnis, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2009, 131, 8720–8721 CrossRef CAS.
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS.
- L. Yi, H. Sun, A. Itzen, G. Triola, H. Waldmann, R. S. Goody and Y.-W. Wu, Angew. Chem., Int. Ed., 2011, 50, 8287–8290 CrossRef CAS.
- J. M. Antos, G.-L. Chew, C. P. Guimaraes, N. C. Yoder, G. M. Grotenbreg, M. W.-L. Popp and H. L. Ploegh, J. Am. Chem. Soc., 2009, 131, 10800–10801 CrossRef CAS.
- N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–884 CrossRef CAS.
- J.-Y. Yang and W. Y. Yang, J. Am. Chem. Soc., 2009, 131, 11644–11645 CrossRef CAS.
- S. Granier, S. Kim, A. M. Shafer, V. R. P. Ratnala, J. J. Fung, R. N. Zare and B. Kobilka, J. Biol. Chem., 2007, 282, 13895–13905 CrossRef CAS.
- (a) H. Neumann, K. Wang, L. Davis, M. Garcia-Alai and J. W. Chin, Nature, 2010, 464, 441–444 CrossRef CAS; (b) B. Wu, Z. Wang, Y. Huang and W. R. Liu, ChemBioChem, 2012, 13, 1405–1408 CrossRef CAS; (c) E. M. Brustad, E. A. Lemke, P. G. Schultz and A. A. Deniz, J. Am. Chem. Soc., 2008, 130, 17664–17665 CrossRef CAS.
- S. Nehring, N. Budisa and B. Wiltschi, PLoS One, 2012, 7, e31992 CAS.
- Y. Santoso, C. M. Joyce, O. Potapova, L. Le Reste, J. Hohlbein, J. P. Torella, N. D. F. Grindley and A. N. Kapanidis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 715–720 CrossRef CAS.
- (a) M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS; (b) Y. Kim, S. O. Ho, N. R. Gassman, Y. Korlann, E. V. Landorf, F. R. Collart and S. Weiss, Bioconjugate Chem., 2008, 19, 786–791 CrossRef CAS; (c) K. L. Holmes and L. M. Lantz, Methods in Cell Biology, Academic Press, 2001, vol. 63, Part A, pp. 185–204 Search PubMed; (d) Q.-F. Li, Y. Yang, A. Maleckis, G. Otting and X.-C. Su, Chem. Commun., 2012, 48, 2704–2706 RSC; (e) V. Chudasama, M. E. B. Smith, F. F. Schumacher, D. Papaioannou, G. Waksman, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 8781–8783 RSC.
- (a) G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 5052–5053 CrossRef CAS; (b) G. J. L. Bernardes, E. J. Grayson, S. Thompson, J. M. Chalker, J. C. Errey, F. El Oualid, T. D. W. Claridge and B. G. Davis, Angew. Chem., Int. Ed., 2008, 47, 2244–2247 CrossRef CAS; (c) J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC; (d) J. M. Chalker, L. Lercher, N. R. Rose, C. J. Schofield and B. G. Davis, Angew. Chem., Int. Ed., 2012, 51, 1835–1839 CrossRef CAS.
- R. Nathani, P. Moody, M. E. B. Smith, R. J. Fitzmaurice and S. Caddick, ChemBioChem, 2012, 13, 1283–1285 CrossRef CAS.
- P. Kuhn, M. Knapp, S. M. Soltis, G. Ganshaw, M. Thoene and R. Bott, Biochemistry, 1998, 37, 13446–13452 CrossRef CAS.
- J.-D. Pedelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79–88 CrossRef CAS.
- S. Hubbard and J. Thornton, Naccess, 1993, University of Manchester, Manchester, U.K, based on B. Lee, F. M. Richards, J. Mol. Biol., 1971, 55, 379–400.
- Images generated using PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC.
Footnote |
| † Electronic supplementary information (ESI) available: LC-MS, ES-MS, deconvoluted spectra and fluorescence emission spectra for all reactions with proteins described herein. Fluorescence emission spectra of superfolder GFP, all the cysteine mutants and their derivatives are given. See DOI: 10.1039/c3sc51333e |
| This journal is © The Royal Society of Chemistry 2013 |