 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed heteroallylation of unactivated alkenes – synthesis of citalopram†
Joanne F. M.
Hewitt
,
Lewis
Williams
,
Pooja
Aggarwal
,
Craig D.
Smith
and
David J.
France
*
WestCHEM, School of Chemistry, University of Glasgow, Joseph Black Building, University Avenue, Glasgow G12 8QQ, UK. E-mail: david.france@glasgow.ac.uk; Fax: +44 (0)141 330 6867; Tel: +44 (0)141 330 4708
First published on 27th June 2013
Abstract
A palladium-catalyzed difunctionalization of unactivated alkenes with tethered nucleophiles is reported. The versatile reaction occurs with simple allylic halides and can be carried out under air. The methodology provides rapid access to a wide array of desirable heterocyclic targets, as illustrated by a concise synthesis of the widely prescribed antidepressantcitalopram.
Introduction
Palladium-catalyzed heterocyclization onto alkenes holds particular importance for synthetic chemists because of the complexity formed, the generally tolerant reaction conditions, and the prevalence of the heterocyclic products in bioactive targets.1 The σ-alkyl Pd(II) intermediate formed by heterocyclization can participate in a range of subsequent transformations including β-hydride elimination (Wacker-type reaction), alkoxycarbonylation, Heck reaction, or protodemetallation.1j In addition to these pathways, when the heterocyclization is preceded by oxidative addition, a reductive elimination can complete the catalytic cycle.2 Recently, strategies have emerged that involve oxidation of the σ-alkyl Pd(II) intermediate formed by heterocyclization (particularly with hypervalent iodine reagents)3 as a means of subsequently forming new C–halogen,4 C–O,5 C–N,6 and C–C6f,7 bonds.When a Pd(II)-induced heterocyclization onto an unactivated alkene is followed by C–C bond formation, the new C–C bond is typically to an sp2-hybridized carbon (e.g.2 → 3, Scheme 1).8,9 Extending this methodology to form sp3–sp3 C–C bonds would constitute a significant advance, in part because increasing the sp3-C content of preclinical drug candidates has been identified as a potential strategy to improve clinical success rates.10 Pd-catalyzed allylation is a powerful method for C–C bond formation that has seen widespread use in the construction of an impressive array of complex targets;11 however, this method has never been coupled to the Pd(II)-induced heterocyclization onto unactivated alkenes. In this communication, we report the catalyzed reaction of unactivated alkenes with tethered oxygen or nitrogen nucleophiles and allylic halides to generate a new fully substituted carbon center and a new sp3–sp3 C–C bond. Furthermore, we demonstrate the utility of this method with a synthesis of the widely prescribed selective serotonin reuptake inhibitor (SSRI) citalopram.
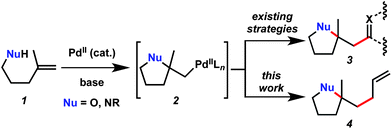 | ||
| Scheme 1 Pd-induced heterocyclization followed by C–C bond formation. | ||
Results and discussion
Optimization
The combination of alkenyl phenol 5 with the commercially available catalyst Pd(hfacac)2 was selected for initial screening of the planned heteroallylation based on precedent in other oxypalladation reactions (Table 1).7b Gratifyingly, benzofuran6 was observed when the reaction with allyl bromide was conducted in THF, though conversion was relatively low after 72 h (entry 1). Solvent screening revealed that ca. 80% conversion could be achieved in PhMe after 16 h (entry 2). Attempting to use alternative Pd catalysts (entries 3–6), or allylic electrophiles (entries 7, 8) resulted in substantially lower conversions. By replacing allyl bromide with allyl chloride,9 full conversion was observed in 6 h (entry 9). Decreasing catalyst loading to 5 mol% resulted in increased reaction time, but full conversion was still observed after 16 h (entry 10).12 It should be noted that the optimized conditions can be conducted under ambient atmosphere using solvent directly as procured.13 From a mechanistic point of view, it is noteworthy that only trace amounts of dihydrobenzofuran 6 formed when using a Pd(0) catalyst (entry 11, vide infra).|
|
|||||
|---|---|---|---|---|---|
| Entrya | Pd cat. (loading) | X | Solvent | Time |
5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6b :6b |
|
a
Reaction conditions: 5 (1 equiv.), allyl-X (5 equiv.), Pd cat., NaHCO3 (2 equiv.), 50 °C, solvent (0.25 M).
b Determined by 1H NMR integration of crude reaction mixtures.
c No reaction. 5:6 20:<1.
|
|||||
| 1 | Pd(hfacac)2 (10 mol%) | Br | THF | 72 h | 2:1 |
| 2 | Pd(hfacac)2 (10 mol%) | Br | PhMe | 16 h | 1:4.3 |
| 3 | Pd(OAc)2 (10 mol%) | Br | PhMe | 22 h | 19:1 |
| 4 | Pd(acac)2 (10 mol%) | Br | PhMe | 22 h | 12:1 |
| 5 | Pd(TFA)2 (10 mol%) | Br | PhMe | 22 h | N. R.c |
| 6 | PdCl2 (10 mol%) | Br | PhMe | 28 h | 1:2 |
| 7 | Pd(hfacac)2 (10 mol%) | OAc | PhMe | 18 h | N. R.c |
| 8 | Pd(hfacac)2 (10 mol%) | I | PhMe | 18 h | 5:1 |
| 9 | Pd(hfacac)2 (10 mol%) | Cl | PhMe | 6 h | <1:20 |
| 10 | Pd(hfacac)2 (5 mol%) | Cl | PhMe | 16 h | <1:20 |
| 11 | Pd2(dba)3 (10 mol%) | Br | PhMe | 18 h | N. R.c |

Substrate scope
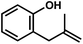
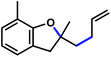
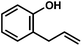
With these optimized conditions in place, we sought to examine the scope of this heteroallylation reaction to make substituted dihydrobenzofurans (Table 2). The unsubstituted case provided the allylation product in 70% isolated yield (entry 1). Substitution ortho to the reacting oxygen and inclusion of electron-withdrawing substituents were tolerated without decrease in yield (entries 2–4).14 Of particular importance is the successful heteroallylation in the presence of an aryl bromide (entry 5), which can serve as a handle for the subsequent introduction of a vast array of functionality. When attempting to utilize a monosubstituted alkene, only trace oxyallylation product was detected, with the major product arising from β-hydride elimination from the putative σ-alkyl Pd(II) intermediate (entry 6, vide infra).15|
|
||||
|---|---|---|---|---|
| Entrya | Substrate | Product | Time | Yieldb |
| a Reaction conditions: substrate (1 equiv.), allyl chloride (5 equiv.), Pd(hfacac)2 (5 mol%), NaHCO3 (2 equiv.), PhMe (0.25 M), 50 °C. b Isolated yield. c Reaction carried out on 0.9 g scale. d Yield not determined. | ||||
| 1 |
|

|
16 h | 70% |
| 2 |

|
|
24 h | 75%c |
| 3 |

|

|
23 h | 74% |
| 4 |

|

|
24 h | 67% |
| 5 |

|

|
16 h | 71% |
| 6 |
|

|
20 h | n.d.d |

Next, we chose to further probe the scope of the heteroallylation reaction by examining the formation of heterocycles other than dihydrobenzofuran (Table 3). Use of the previously optimized reaction conditions with a benzylic alcohol led to a relatively low overall yield (entry 1).16 Fortunately, recourse to allyl bromide resulted in a cleaner reaction profile, with the desired isobenzofuran being obtained in 77% isolated yield (entry 2). Steric hindrance about the unactivated alkene did not prove to be an impediment to the hetroallylation reaction (entry 4). The dihydroisobenzopyran ring system could also be assembled using this methodology (entry 5).17 Use of a substrate containing an unprotected hydroxyl group that was not involved in the heterocyclization resulted in low conversion with allyl bromide; however, using allyl chloride for this substrate resulted in a synthetically viable yield (entries 7, 8). Secondary and tertiary alcohols, as well as benzoic acids also performed well in the reaction (entries 9–14). It should be highlighted that substitution on the unactivated alkene can include sp3-C (including a sterically encumbering t-Bu group), or an aromatic ring.18 Once again, when attempting the oxyallylation with a monosubstituted alkene, cyclization followed by β-hydride elimination occurred in relatively low conversion, with none of the allylated lactone being observed (entry 15).15
|
a
Reaction conditions: substrate (1 equiv.), allyl-X (5 equiv.), Pd(hfacac)2 (5 mol%), NaHCO3 (2 equiv.), PhMe (0.25 M), 50 °C. bIsolated yield except where noted. cYield at full conversion based on 1H NMR integration, compound contaminated with additional impurities. dAn additional 5 mol% catalyst was added. eCarried out in d8-toluene; yield based on 1H NMR integration vs. internal standard. fd.r. = 1.4:1 see ESI for all assignments. gd.r. = 1.3:1. hd.r. = 1.5:1. iLow conversion to a mixture of products, see ESI. |
|---|

|
We then attempted to extend the method to an aminoallylation by cyclization of tosyl amides onto unactivated alkenes (Scheme 2). These efforts were initially plagued by low conversion on gem-disubstituted alkene precursors even at prolonged reaction times with 10 mol% catalyst loading. However, we were successfully able to access the isoquinolone (8b) and pyrrolopyrazinone (8c) ring systems from monosubstituted alkene precursors without significant β-hydride elimination.15 Modification of the base to KH2PO4 was required for these aminocyclizations in order to suppress N-allylation.
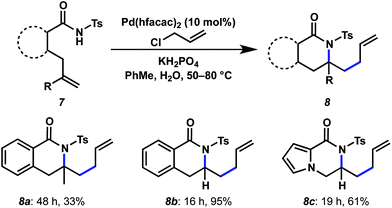 | ||
| Scheme 2 Aminoallylation substrates and conditions. | ||
Mechanistic hypothesis
Although the precise mechanism for this heteroallylation reaction is yet to be fully elucidated, we have carried out preliminary studies designed to test the possible intermediacy of a π-allyl species. An experiment using dideuteroallyl bromide labelled at the allylic position resulted in exclusive formation of a product with two vinyldeuterons (Scheme 3).15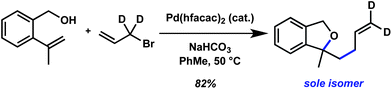 | ||
| Scheme 3 Deuterium labelling study. | ||
Furthermore, while not definitive, the lack of reactivity using Pd2dba3, coupled with the fact that precipitation of Pd(0) was not observed during the course of the reactions and the tolerance of the process for aryl halides (which might have undergone oxidative addition to an in situ-generated Pd(0) species) suggests that Pd(0) intermediates are not involved.13 These factors lead us to propose the following catalytic cycle (Scheme 4): Pd(II)-induced heterocyclization (9 → 10) might be followed by carbopalladation of the allyl halide to generate Pd(II)-alkyl complex 11.1,19 Subsequent β-halide elimination (which has been shown to occur more rapidly than β-hydride elimination in several systems9,20) would then afford heteroallylation product 12, while releasing the Pd(II) catalyst.21 This mechanistic interpretation is consistent with results obtained in related cyclizations of allenes.9e,g,h
 | ||
| Scheme 4 Postulated catalytic cycle. | ||
Synthesis of citalopram
In order to demonstrate the utility of the heteroallylation methodology for the preparation of bioactive heterocycles, and further probe substrate scope, a synthesis of the SSRI citalopram was performed (Scheme 5).22,23 The reaction of p-fluorophenyl Grignard reagent14 with commercially available cyanophthalide 13 is known from the patent literature.23c,f Subsequent Wittig reaction provided the requisite alkenyl alcohol15. The pivotal Pd-catalyzed oxyallylation proceeded in good yield to furnish isobenzofuran16. Conversion of terminal alkene16 to citalopram was readily achieved by one-pot dihydroxylation–oxidative cleavage,24 followed by reductive amination. | ||
| Scheme 5 Synthesis of citalopram using oxyallylation. | ||
Conclusions
In summary, we have developed a versatile catalytic heteroallylation reaction of unactivated alkenes to form heterocycles that are prevalent in a plethora of targets including natural products and bioactive compounds. The process forms a fully substituted carbon center and a new sp3–sp3 C–C bond in a single step, and occurs using a commercially available catalyst under operationally convenient conditions (e.g. under air). The orthogonality of the newly described heteroallylation to standard Pd(0)-catalyzed processes (as illustrated by the tolerance of the reaction for aryl halides, and lack of reactivity with a Pd(0) catalyst) clearly shows the potential for this method to be used in accessing complex targets. Phenols, alcohols, carboxylic acids and tosyl amides are all competent nucleophiles for this transformation, which we have also shown can be employed in a synthesis of the bioactive heterocycle citalopram. Ongoing investigations in our group seek to further expand the scope of the reaction, including ligand-based control of enantioselectivity, and explore the mechanism underpinning the process.Acknowledgements
The authors are grateful to the EPSRC, WestCHEM, the University of Glasgow, Pfizer, and AstraZeneca for financial support.Notes and references
- (a) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, Wiley, New York, 2002, vol. 2 Search PubMed; (b) G. Balme, D. Bouyssi, T. Lomberget and N. Monteiro, Synthesis, 2003, 2115 CrossRef CAS; (c) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285 CrossRef CAS; (d) L. F. Tietze, H. Ila and H. P. Bell, Chem. Rev., 2004, 104, 3453 CrossRef CAS; (e) A. Minatti and K. Muñiz, Chem. Soc. Rev., 2007, 36, 1142 RSC; (f) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS; (g) J. P. Wolfe, Eur. J. Org. Chem., 2007, 571 CrossRef CAS; (h) J. P. Wolfe, Synlett, 2008, 2913 CrossRef CAS; (i) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 4083 RSC; (j) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS; (k) D. M. Schultz and J. P. Wolfe, Synthesis, 2012, 351 CAS; (l) J. P. Wolfe, Angew. Chem., Int. Ed., 2012, 51, 10224 CrossRef CAS.
- (a) M. F. Semmelhack and W. R. Epa, Tetrahedron Lett., 1993, 34, 7205 CrossRef CAS; (b) J. P. Wolfe and M. A. Rossi, J. Am. Chem. Soc., 2004, 126, 1620 CrossRef CAS; (c) J. E. Ney and J. P. Wolfe, Angew. Chem., Int. Ed., 2004, 43, 3605 CrossRef; (d) R. Lira and J. P. Wolfe, J. Am. Chem. Soc., 2004, 126, 13906 CrossRef CAS; (e) J. E. Ney, M. B. Hay, Q. Yang and J. P. Wolfe, Adv. Synth. Catal., 2005, 347, 1614 CrossRef CAS; (f) S. Hayashi, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2009, 131, 2052 CrossRef CAS; (g) S. Hayashi, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2009, 48, 7224 CrossRef CAS; (h) J. D. Neukom, N. S. Perch and J. P. Wolfe, J. Am. Chem. Soc., 2010, 132, 6276 CrossRef CAS; (i) D. N. Mai and J. P. Wolfe, J. Am. Chem. Soc., 2010, 132, 12157 CrossRef CAS; (j) S. G. Newman and M. Lautens, J. Am. Chem. Soc., 2011, 133, 1778 CrossRef CAS; (k) H. Liu, C. Li, D. Qiu and X. Tong, J. Am. Chem. Soc., 2011, 133, 6187 CrossRef CAS; (l) S. Nicolai and J. Waser, Org. Lett., 2011, 13, 6324 CrossRef CAS; (m) S. G. Newman, J. K. Howell, N. Nicolaus and M. Lautens, J. Am. Chem. Soc., 2011, 133, 14916 CrossRef CAS; (n) D. C. Koester, M. Kobayashi, D. B. Werz and Y. Nakao, J. Am. Chem. Soc., 2012, 134, 6544 CrossRef CAS; (o) B. A. Hopkins and J. P. Wolfe, Angew. Chem., Int. Ed., 2012, 51, 9886 CrossRef CAS; (p) S. Nicolai, R. Sedigh-Zadeh and J. Waser, J. Org. Chem., 2013, 78, 3783 CrossRef CAS; (q) N. Sun, Y. Li, G. Yin and S. Jiang, Eur. J. Org. Chem., 2013, 13, 2541 CrossRef.
- (a) N. R. Deprez and M. S. Sanford, Inorg. Chem., 2007, 46, 1924 CrossRef CAS; (b) K. Muñiz, Angew. Chem., Int. Ed., 2009, 48, 9412 CrossRef; (c) L.-M. Xu, B.-J. Li, Z. Yang and Z.-J. Shi, Chem. Soc. Rev., 2010, 39, 712 RSC; (d) P. Sehnal, R. J. K. Taylor and I. J. S. Fairlamb, Chem. Rev., 2010, 110, 824 CrossRef CAS; (e) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS.
- (a) M. R. Manzoni, T. P. Zabawa, D. Kasi and S. R. Chemler, Organometallics, 2004, 23, 5618 CrossRef CAS; (b) A. Lei, X. Lu and G. Liu, Tetrahedron Lett., 2004, 45, 1785 CrossRef CAS; (c) F. E. Michael, P. A. Sibbald and B. M. Cochran, Org. Lett., 2008, 10, 793 CrossRef CAS; (d) T. A. Doroski, M. R. Cox and J. B. Morgan, Tetrahedron Lett., 2009, 50, 5162 CrossRef CAS; (e) T. Wu, G. Yin and G. Liu, J. Am. Chem. Soc., 2009, 131, 16354 CrossRef CAS.
- (a) E. J. Alexanian, C. Lee and E. J. Sorensen, J. Am. Chem. Soc., 2005, 127, 7690 CrossRef CAS; (b) Y. Li, D. Song and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 2962 CrossRef CAS; (c) D. V. Liskin, P. A. Sibbald, C. F. Rosewall and F. E. Michael, J. Org. Chem., 2010, 75, 6294 CrossRef CAS; (d) Y.-B. Kang and L. H. Gade, J. Am. Chem. Soc., 2011, 133, 3658 CrossRef CAS.
- (a) J. Streuff, C. H. Hövelmann, M. Nieger and K. Muñiz, J. Am. Chem. Soc., 2005, 127, 14586 CrossRef CAS; (b) L. V. Desai and M. S. Sanford, Angew. Chem., Int. Ed., 2007, 46, 5737 CrossRef CAS; (c) K. Muñiz, J. Am. Chem. Soc., 2007, 129, 14542 CrossRef; (d) K. Muñiz, C. H. Hövelmann and J. Streuff, J. Am. Chem. Soc., 2008, 130, 763 CrossRef; (e) P. A. Sibbald and F. E. Michael, Org. Lett., 2009, 11, 1147 CrossRef CAS; (f) P. A. Sibbald, C. F. Rosewall, R. D. Swartz and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 15945 CrossRef CAS; (g) E. L. Ingalls, P. A. Sibbald, W. Kaminsky and F. E. Michael, J. Am. Chem. Soc., 2013, 135, 8854 CrossRef CAS.
- (a) C. F. Rosewall, P. A. Sibbald, D. V. Liskin and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 9488 CrossRef CAS; (b) S. Nicolai, S. Erard, D. Fernández González and J. Waser, Org. Lett., 2010, 12, 384 CrossRef CAS; (c) S. Nicolai, C. Piemontesi and J. Waser, Angew. Chem., Int. Ed., 2011, 50, 4680 CrossRef CAS.
- Other methods of heterocyclization onto unactivated alkenes with concomitant sp3–sp3 C–C bond formation include use of organomercurials (a–d), radical cyclizations (e–h) including Co-catalyzed reactions (i–k), Pd-catalyzed reactions with other subsequent processes (2p, l–o), or other related pathways (p, q): (a) B. Giese and K. Heuck, Chem. Ber., 1981, 114, 1572 CrossRef CAS; (b) S. Danishefsky, E. Taniyama and R. R. Webb II, Tetrahedron Lett., 1983, 24, 11 CrossRef CAS; (c) W. Carruthers, M. J. Williams and M. T. Cox, J. Chem. Soc., Chem. Commun., 1984, 1235 RSC; (d) D. R. Adams, W. Carruthers, M. J. Williams and P. J. Crowley, J. Chem. Soc., Perkin Trans. 1, 1989, 1507 RSC; (e) H. Yorimitsu, K. Wakabayashi, H. Shinokubo and K. Oshima, Tetrahedron Lett., 1999, 40, 519 CrossRef CAS; (f) H. Yorimitsu, K. Wakabayashi, H. Shinokubo and K. Oshima, Bull. Chem. Soc. Jpn., 2001, 74, 1963 CrossRef CAS; (g) T. W. Liwosz and S. R. Chemler, J. Am. Chem. Soc., 2012, 134, 2020 CrossRef CAS; (h) R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 12462 CrossRef CAS; (i) D. Schuch, P. Fries, M. Dönges, B. Menéndez Pérez and J. Hartung, J. Am. Chem. Soc., 2009, 131, 12918 CrossRef CAS; (j) P. Fries, D. Halter, A. Kleinschek and J. Hartung, J. Am. Chem. Soc., 2011, 133, 3906 CrossRef CAS; (k) P. Fries, M. K. Müller and J. Hartung, Org. Biomol. Chem., 2013, 11, 2630 RSC; (l) M. A. Arai, M. Kuraishi, T. Arai and H. Sasai, J. Am. Chem. Soc., 2001, 123, 2907 CrossRef CAS; (m) K.-T. Yip, M. Yang, K.-L. Law, N.-Y. Zhu and D. Yang, J. Am. Chem. Soc., 2006, 128, 3130 CrossRef CAS; (n) K.-T. Yip, N.-Y. Zhu and D. Yang, Org. Lett., 2009, 11, 1911 CrossRef CAS; (o) B. M. Trost and J. Rey, Org. Lett., 2012, 14, 5632 CrossRef CAS; (p) S. Kumar, J.-C. P. Helt, J. Autschbach and M. R. Detty, Organometallics, 2009, 28, 3426 CrossRef CAS; (q) K. Komeyama, Y. Kouya, Y. Ohama and K. Takaki, Chem. Commun., 2011, 47, 5031 RSC.
- Heterocyclizations involving allylic electrophiles have been demonstrated for alkynes (a, b) and allenes (c–h): (a) Y. Wakabayashi, Y. Fukuda, H. Shiragami, K. Utimoto and H. Nozaki, Tetrahedron, 1985, 41, 3655 CrossRef CAS; (b) Y. Fukuda, H. Shiragami, K. Utimoto and H. Nozaki, J. Org. Chem., 1991, 56, 5816 CrossRef CAS; (c) M. Kimura, K. Fugami, S. Tanaka and Y. Tamaru, J. Org. Chem., 1992, 57, 6377 CrossRef CAS; (d) S. Ma and L. Li, Org. Lett., 2000, 2, 941 CrossRef CAS; (e) S. Ma and W. Gao, J. Org. Chem., 2002, 67, 6104 CrossRef CAS; (f) S. Ma, F. Yu and W. Gao, J. Org. Chem., 2003, 68, 5943 CrossRef CAS; (g) S. Ma and Z. Yu, J. Org. Chem., 2003, 68, 6149 CrossRef CAS; (h) B. Alcaide, P. Almendros, R. Carrascosa and T. Martínez del Campo, Chem.–Eur. J., 2010, 16, 13243 CrossRef CAS.
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS; (b) T. J. Ritchie, S. J. F. Macdonald, R. J. Young and S. D. Pickett, Drug Discovery Today, 2011, 16, 164 CrossRef CAS; (c) A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114 CrossRef CAS.
- (a) R. F. Heck, J. Am. Chem. Soc., 1968, 90, 5531 CrossRef CAS; (b) R. C. Larock, J. C. Bernhardt and R. J. Driggs, J. Organomet. Chem., 1978, 156, 45 CrossRef CAS; (c) J. Tsuji, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, WILEY-VCH, New York, 2002, vol. 2, pp. 1669–1767 Search PubMed; (d) B. M. Trost and D. L. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS; (e) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS; (f) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS.
- Other combinations of catalyst, ligand, base, and solvent were less successful.
- Carrying out the reaction under Ar using degassed solvent did not significantly impact conversion, suggesting that O2 is not involved in the process.
- A side product resulting from heterocyclization followed by Heck reaction onto the product alkene was isolated from the reaction shown in entry 2 accounting for the majority of the mass balance. See ESI for further details.†.
- See ESI for details.†.
- No trace of aldehyde resulting from Pd(II)-promoted alcohol oxidation was observed for any reaction in Table 3. See T. Nishimura and S. Uemura, Synlett, 2004, 201 CAS and references therein for examples of Pd(II)-promoted alcohol oxidation.
- Extension to ring sizes larger than 6-membered resulted in markedly decreased yields.
- Substitution on the allyl component is less well tolerated under the present conditions. For example, reaction of a benzylic alcohol with 1-chloro-3-methyl-2-butene provided only a 20% isolated yield of the oxyallylation product after 5 d at 50 °C. Use of 1,2-disubstituted alkenes leads to β-hydride elimination.
- See ref. 2a and 8l as well as: (a) L. S. Hegedus, G. F. Allen and D. J. Olsen, J. Am. Chem. Soc., 1980, 102, 3583 CrossRef CAS; (b) R. C. Larock and N. H. Lee, J. Am. Chem. Soc., 1991, 113, 7815 CrossRef CAS; (c) L. F. Tietze, K. M. Sommer, J. Zinngrebe and F. Stecker, Angew. Chem., Int. Ed., 2005, 44, 257 CrossRef CAS.
- See ref. 11a and b, as well as: (a) X. Lu, Top. Catal., 2005, 35, 73 CrossRef; (b) H. Zhao, A. Ariafard and Z. Lin, Organometallics, 2006, 25, 812 CrossRef CAS; (c) J. Le Bras and J. Muzart, Tetrahedron, 2012, 68, 10065 CrossRef CAS.
- An alternative pathway that proceeds via a Pd(IV)-allyl complex may also be possible. See: (a) P. K. Byers and A. J. Canty, J. Chem. Soc., Chem. Commun., 1988, 639 RSC; (b) R. Guo, J. L. Portscheller, V. W. Day and H. C. Malinakova, Organometallics, 2007, 26, 3874 CrossRef CAS.
- Clinical use: M. B. Keller, J. Clin. Psychiatry, 2000, 61, 896 CrossRef CAS.
- Existing syntheses of citalopram: (a) K. P. Bøgesø and A. S. Toft, U.S. Pat. 4,136,193, 1979; (b) K. P. Bøgesø, U.S. Pat. 4,650,884, 1987; (c) H. Petersen, K. P. Bøgesø and M. Bech Sommer, WO Pat. 98/19511, 1998; (d) H. Petersen, P. Bregnedal and K. P. Bøgesø, WO Pat. 98/19512, 1998; (e) H. Petersen, WO Pat. 98/19513, 1998; (f) M. H. Rock, H. Petersen and P. Ellegaard, WO Pat. 00/12044, 2000; (g) R. Vedantham, P. V. N. K. V. Vetukuri, A. Boini, M. Khagga and R. Bandichhor, Org. Process Res. Dev., 2013, 17, 798 CrossRef CAS.
- (a) R. Pappo, D. S. Allen Jr, R. U. Lemieux and W. S. Johnson, J. Org. Chem., 1956, 21, 478 CrossRef CAS; (b) V. VanRheenen, R. C. Kelly and D. Y. Cha, Tetrahedron Lett., 1976, 17, 1973 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and compound characterization data. See DOI: 10.1039/c3sc51222c |
| This journal is © The Royal Society of Chemistry 2013 |