 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamic and bio-orthogonal protein assembly along a supramolecular polymer†
Katja
Petkau-Milroy‡
a,
Dana A.
Uhlenheuer‡
a,
A. J. H.
Spiering
b,
Jef A. J. M.
Vekemans
b and
Luc
Brunsveld
*a
aLaboratory of Chemical Biology and Institute for Complex Molecular Systems, Department of Biomedical Engineering, Eindhoven University of Technology, Den Dolech 2, 5612 AZ Eindhoven, The Netherlands. E-mail: l.brunsveld@tue.nl; Fax: +31 40 2478367
bLaboratory of Macromolecular and Organic Chemistry, Department of Chemistry and Chemical Engineering, Eindhoven University of Technology, Den Dolech 2, 5612 AZ Eindhoven, The Netherlands
First published on 26th April 2013
Abstract
Dynamic protein assembly along supramolecular columnar polymers has been achieved through the site-specific covalent attachment of different SNAP-tag fusion proteins to self-assembled benzylguanine-decorated discotics. The self-assembly of monovalent discotics into supramolecular polymers creates a multivalent, bio-orthogonal and self-regulating framework for protein assembly. The intrinsic reversibility of supramolecular interactions results in reorganization and exchange of building blocks allowing for dynamic intermixing of protein-functionalized discotics between different self-assembled polymers, leading to self-optimization of protein arrangement and distance as evidenced by efficient energy transfer between fluorescent proteins.
Introduction
The assembly of proteins in cells and on cell membranes is essential for a diverse range of fundamental biological processes including gene transcription, signal transduction and the regulation of metabolic pathways.1–3 The underlying design principles of these natural protein assemblies such as well-defined spatial organization and shape, induced proximity, and multivalency have inspired the design of synthetic scaffolds for protein assembly based on small molecules,4–7 dendrimers,8–10 and natural and synthetic polymers,11–17 which exemplify many of these beneficial features with applications in diagnostics and nanotechnology. Another key element of natural protein assemblies is their reversibility which serves as a self-regulatory mechanism, for example during gene transcription, cell motility and cell division.18 The dynamic assembly and disassembly of the gene transcription machinery or of actin filaments renders the cellular processes susceptible to changes. Synthetic systems which possess an inherently dynamic framework for protein assembly will provide a point of entry to synthetic biological architectures with dynamic properties and functions that mimic their biologically inspired counterparts.12Due to their responsive nature, supramolecular assemblies offer a promising synthetic platform for dynamic chemistry,19–21 bridging the gap to dynamic biological systems.22 Ease of assembly, modularity, tunability and the potential to incorporate multiple copies of different active units are further advantages of self-assembled systems ranging from micellar-like architectures to completely synthetic nanostructures.23–25 Recently, we have reported a class of auto-fluorescent C3-symmetrical amphiphilic discotics which instantly assemble into supramolecular columnar stacks in water.26 These discotics have been functionalized with different bio-active small molecules, such as sugars or biotin.27,28 Studies with trivalent functionalized discotics and multivalent proteins such as streptavidin and concanavalin A showed the binding of several proteins to these self-assembled discotics. The multivalent nature of these protein–ligand interactions used by us and others,5–7,15,28 however, limits full molecular control over the number of proteins appended and hampers investigations into the dynamics of the supramolecular protein assembly.
Herein we present the site-specific, bio-orthogonal, and covalent conjugation of single proteins to an intrinsically monovalent discotic molecule functionalized with one O6-benzylguanine moiety (Fig. 1a). Separate conjugation of two types of fluorescent proteins (yellow and cyan fluorescent) allows the self-assembling discotic scaffold to form supramolecular architectures that can act as a multivalent and dynamic protein-assembly platform (Fig. 1b). The dynamic nature of the supramolecular polymer allows the fluorescent proteins to come together leading to efficient energy transfer, but also to dynamically adjust their localization upon building block exchange and density tuning, resulting in optimized and adaptable energy transfer (Fig. 1c).
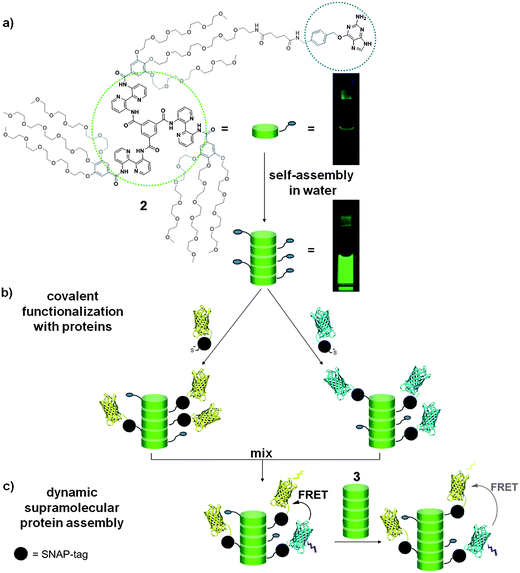 | ||
| Fig. 1 (a) Structure of the O-benzylguanine (BG) discotic 2 carrying a single moiety for conjugation to SNAP-tag fusion proteins. In water these discotics self-assemble to form auto-fluorescent columnar stacks, displaying moieties for conjugation at their periphery. The photographs show 10 μM solutions of 2 in CH2Cl2 (top) and in water (bottom) excited with UV light (λex = 350 nm). (b) Site-selective covalent functionalization of the supramolecular polymer with a SNAP-tag fused to cyan and yellow fluorescent protein (CFP and YFP). (c) Dynamic intermixing of supramolecular protein assemblies results in efficient Förster resonance energy transfer (FRET) between CFP and YFP. Intercalation of inert-discotic 3 allows tuning of the distance between the fluorescent proteins. | ||
Results and discussion
Design and synthesis
Covalent, monovalent and site-specific protein conjugation was sought after to study the dynamics of supramolecular protein assembly. Covalent attachment circumvents protein dissociation and facilitates the characterization of protein-conjugates. Monovalent and site-specific attachment ensures reproducibility and a well-defined stoichiometric composition. Therefore, a discotic supramolecular building block mono-functionalized with O6-benzylguanine (2), a reactive functionality for bio-orthogonal protein conjugation, was designed, offering a non-sterically hindered general platform for the covalent attachment of diverse proteins. This intrinsically monovalent building block should, only upon self-assembly, generate a multivalent polymer displaying several proteins in mutual proximity, but with dynamic adaptability.Selective mono-functionalization with O6-benzylguanine required the synthesis of a C2-symmetric discotic molecule featuring a single amine at one of its nine peripheral oligo(ethylene oxide) chains. Desymmetrization was achieved through conversion of acid 4 into the corresponding dichloride and subsequent amidation with 5 (Scheme 1). The saponification of the resulting ester 6 was performed using LiOH in water to prevent hydrolysis of the aromatic amides. A mild chlorination agent, 1-chloro-N,N,2-trimethylpropenylamine,29 was used to convert the resulting acid into an acid chloride without affecting the secondary amide bonds of the molecule. Amidation with 7 bearing a single Boc-protected amine and subsequent deprotection afforded the desired desymmetrized monoamine discotic 1 in high yield. O6-(4-glutarylamidomethyl)benzylguanine30 was coupled to this amine-functionalized discotic 1 in dimethylacetamide using HBTU and DIPEA and the excess of O6-(4-glutarylamidomethyl)benzylguanine was removed by size exclusion chromatography to give pure O-benzylguanine-discotic (BG-discotic) 2 in 90% yield.
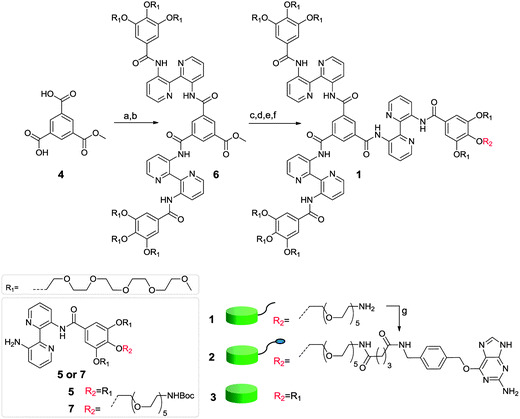 | ||
| Scheme 1 Synthesis of benzylguanine-discotic 2 and structure of the inert-discotic 3. Reagents and conditions: (a) (COCl)2, DMF, CH2Cl2, 0 °C, 1 h, quant., (b) 5, NEt3, CH2Cl2, RT, 18 h, 52%, (c) (i) LiOH, H2O, 85 °C, 16 h, (ii) aq. oxalic acid, 85%, (d) 1-chloro-N,N,2-trimethylpropenylamine, RT, 2 h, quant., (e) 7, NEt3, CH2Cl2, RT, 18 h, 77%, (f) TFA, CH2Cl2, RT, 1 h, quant. and (g) O6-(4-glutarylamidomethyl)benzylguanine, HBTU, DIPEA, DMA, RT, 4 h, 90%. | ||
Functionalization of discotics with fluorescent proteins
The specific reaction of O6-benzylguanine with the SNAP-tag,31,32 a genetically encoded protein tag which can be fused to the protein of interest, provides an entry for site-selective covalent attachment of proteins to discotic 2. This self-labelling tag offers entry to highly selective and single protein labelling and, in contrast to widely applied bioconjugation techniques such as thiol–maleimide chemistry, enables labelling in complex mixtures, for example for future cellular work.33 Both, yellow and cyan fluorescent proteins were therefore expressed and purified as SNAP-tag fusion proteins (YFP-SNAP and CFP-SNAP, respectively).34 The proteins were conjugated to the self-assembled BG-discotic 2 in phosphate buffer at micromolar concentrations (see ESI†). The auto-fluorescence of the discotics in the self-assembled state (λexc = 340 nm, λem = 510 nm, dashed line in Fig. 2a) makes these supramolecular polymers ideal FRET donors for the YFP protein (λexc = 515 nm) and should allow on-line monitoring of the functionalization of 2via fluorescence spectroscopy. Fluorescence spectroscopy measurements revealed a time-dependent increase of the YFP emission at 527 nm during the protein conjugation reaction upon excitation of the discotics (Fig. 2a). Incubation of YFP-SNAP with inert-discotic 3, not featuring a reactive O6-benzylguanine handle, did not result in FRET (Fig. 2b). These results prove that the observed energy transfer in case of the BG-discotic 2 with YFP-SNAP is specifically due to the covalent protein attachment. Attachment of CFP-SNAP, not being an efficient FRET acceptor for the discotic, did not lead to changes in the self-assembly induced emission spectra of 2. This indicates that the protein functionalization of the discotics does not negatively affect their self-assembly behaviour (see Fig. S9, ESI†). The supramolecular polymer thus ably displays the proteins at its periphery, while efficiently enabling interplay of the chromophores of the polymer with that of the appended proteins.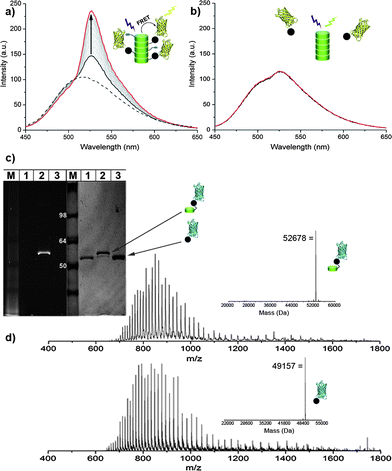 | ||
| Fig. 2 (a and b) Emission spectra of a mixture of YFP-SNAP (1 μM) with either BG-discotic 2 (3 μM, (a)) or inert-discotic 3 (3 μM, (b)) at t = 0 (black) and t = 24 h (red) measured at 20 °C. The dashed line shows the emission of the discotic before the addition of protein; λexc = 340 nm. (c) SDS-PAGE of CFP-SNAP (1), and mixtures of CFP-SNAP with either BG-discotic 2 (2) or inert-discotic 3 (3) after incubation for 3 h at 37 °C. Left: UV illumination, right: Coomassie blue staining. (d) ESI-MS spectra and deconvoluted mass of CFP-discotic conjugate (top) and of CFP-SNAP (bottom).35 | ||
The covalent and single protein conjugation to the discotics was further confirmed by SDS-PAGE and LC-MS. The SDS gel of a ligation mixture of CFP-SNAP with 2, for example, shows a new band at a higher molecular weight, corresponding to the protein covalently linked to 2 (Fig. 2c). Only this band features fluorescence when excited at 350 nm, resulting from the conjugated discotic. No change in protein size and fluorescence was observed upon incubation with the inert discotic 3 (Fig. 2c). LC-MS analysis of the reaction products confirmed the correct corresponding masses of the discotic functionalized proteins (Fig. 2d and S5, ESI†).
Supramolecular assembly of multiple proteins
The self-assembling multivalency25,36 of these intrinsically monovalent discotics bodes well for the display of different proteins along the same supramolecular polymer. The FRET-pair proteins YFP- and CFP-SNAP were therefore simultaneously ligated to a solution of 2. For this, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of YFP-SNAP and CFP-SNAP was added to the self-assembled BG-discotic 2 and in a control experiment to the self-assembled inert-discotic 3. Only when CFP is in close proximity to YFP the excited-state energy of CFP is transferred to YFP, resulting in an increase in YFP fluorescence intensity and a decrease in CFP fluorescence intensity. Only the supramolecular polymer decorated with the reactive O6-benzylguanine handles featured energy transfer from CFP to YFP, resulting in an effective FRET ratio of 0.68 (Fig. 3). The control mixture with the inert supramolecular polymers featured only CFP fluorescence (Fig. 3, black line). These results show that the intrinsically monovalent BG-discotics 2, upon self-assembly, offer a multivalent scaffold for protein attachment and assembly, leading to mutual proximity of different proteins along the supramolecular polymer.
:1 mixture of YFP-SNAP and CFP-SNAP was added to the self-assembled BG-discotic 2 and in a control experiment to the self-assembled inert-discotic 3. Only when CFP is in close proximity to YFP the excited-state energy of CFP is transferred to YFP, resulting in an increase in YFP fluorescence intensity and a decrease in CFP fluorescence intensity. Only the supramolecular polymer decorated with the reactive O6-benzylguanine handles featured energy transfer from CFP to YFP, resulting in an effective FRET ratio of 0.68 (Fig. 3). The control mixture with the inert supramolecular polymers featured only CFP fluorescence (Fig. 3, black line). These results show that the intrinsically monovalent BG-discotics 2, upon self-assembly, offer a multivalent scaffold for protein attachment and assembly, leading to mutual proximity of different proteins along the supramolecular polymer.
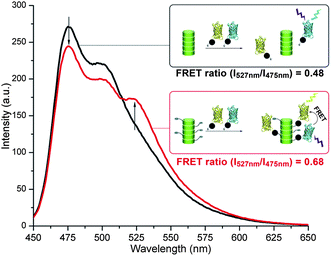 | ||
| Fig. 3 Emission spectra (λexc = 410 nm) of 1:1 mixtures of YFP-SNAP (1 μM) and CFP-SNAP (1 μM) incubated for 3 h at 37 °C with either the BG-discotic 2 (20 μM; red trace) or inert-discotic 3 (20 μM; black trace). | ||
Dynamic intermixing
The modular nature of these supramolecular polymers should enable the generation of heterovalent supramolecular polymers via post-assembly intermixing of differently functionalized discotic building blocks (for concept see Fig. 1c).37 The exchange of protein-conjugated discotics between different supramolecular polymers would rearrange the two different proteins (CFP and YFP), leading to mutual proximity. Thus dynamic intermixing experiments can easily be followed in real time using FRET experiments. Supramolecular protein polymers decorated with either YFP or CFP were first prepared by separate attachment of the individual proteins to the self-assembled BG-discotic 2. Subsequently these solutions were mixed and the appearance of FRET was followed over time (Fig. 4). A strong time-dependent increase in FRET is observed after mixing the two supramolecular protein assemblies.38,39 This evidences that the discotics (monomer = 3.5 kDa) decorated with large proteins (50 kDa) exchange between the supramolecular polymers. As a control the same protein mixtures were prepared using inert-discotic 3 and this mixture did not feature any increase in latent FRET. This also proves the absence of unspecific protein aggregation overtime as well as the requirement of covalent attachment of proteins to the discotic monomers for an increase in energy transfer upon dynamic intermixing. The monomer exchange is rapid; after 10 min the half-maximal increase in FRET ratio is reached. The data is fitted best with a double exponential fit (Fig. S13, ESI†) indicating the presence of both a fast and a slower exchange process, the mechanism behind this is still under investigation.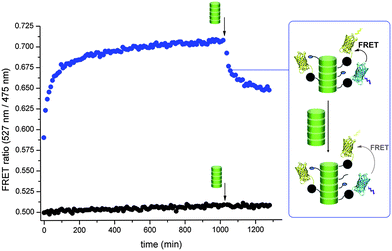 | ||
| Fig. 4 FRET ratios (λexc = 410 nm) of just mixed samples of 2 separately ligated to YFP-SNAP and CFP-SNAP (blue) and of just mixed control samples of 3 with YFP-SNAP and CFP-SNAP (black). Final concentration of discotics is 20 μM and of proteins 1 μM each. The intermixing of the two protein-functionalized supramolecular polymers leads to a heterovalent supramolecular protein polymer containing both CFP and YFP and results in an increase in FRET ratio over time. Arrow indicates addition of 20 μM of 3. For corresponding fluorescence spectra and the non-corrected FRET-ratios see ESI.† | ||
Tuning of protein density
The supramolecular nature of the polymers, in principle allows inserting additional monomers into the main-chain. Therefore, inert-discotic 3 was added to the mixed supramolecular polymer assemblies of YFP and CFP (Fig. 4, arrow). When intermixing with the already present supramolecular protein assemblies, the inert-discotics should act as spacers leading to an increased distance between the fluorescent proteins (for concept see Fig. 1c). Indeed, a rapid decrease in energy transfer was observed upon addition of 3. Additionally, the distance between the proteins and with it the decrease in FRET ratio could be tuned via the addition of different concentrations of 3 (see Fig. S16, ESI†). These results show that in contrast to the typically applied strategy for the generation of heterovalent supramolecular polymers – pre-mixing different building blocks prior to self-assembly40,41 – heterovalent structures based on discotics can as well be formed and fine-tuned after self-assembly.Conclusions
This proof-of-principle work using fluorescent proteins as model proteins showed the possibility to generate adaptable, hetero- and multivalent protein assemblies using supramolecular polymers based on monovalent discotics. A synthetic protocol has been devised which provides access to monovalent discotic molecules with reactive functionalities for protein conjugation, which self-assemble into supramolecular polymers in water. This supramolecular scaffold allows for site-specific covalent attachment of proteins resulting, for example, in energy transfer from the auto-fluorescent discotic building blocks to the attached YFP proteins. When self-assembled, the intrinsically monovalent discotics display multiple proteins along the assembled supramolecular polymer. The conjugation of two different proteins, the FRET-pair CFP and YFP, to this supramolecular polymer then brings these proteins in mutual proximity, as observed by FRET. The adaptable nature of the supramolecular assembly at the same time creates a dynamic framework which is able to assemble proteins in a self-regulating manner. These self-assembled protein-conjugated discotics are fully capable of intermixing, which allows the formation of hetero-functionalized supramolecular protein-conjugated polymers and tuning of protein distance. We envision that such synthetic supramolecular polymers can bridge the gap between synthetic and biological systems, thereby creating dynamic multi- and heterovalent protein assemblies with a responsive nature. The use of other types of proteins will involve other means of monitoring dynamic assembly, for example via the enzymatic activity of assembled split-enzymes or enzyme arrays. Also, further investigations into the hierarchical assembly process, with for example (cryo)electron or atomic force microscopy, will aid to generate dynamic miniaturized protein arrays of different length, via tuning of the discotic concentration. At the same time, the possibility to generate adaptable heterovalent supramolecular polymers after the self-assembly will not only allow to dynamically address protein–protein interactions, but it will as well facilitate the generation and optimization of heterovalent structures as required for applications such as (multimodal) targeted imaging or tissue engineering.42Acknowledgements
The research leading to these results has received funding from the Ministry of Education, Culture and Science (Gravity program 024.001.035), from ERC grant 204554 – SupraChemBio and CTMM – 03O-201 Mammoth.Notes and references
- T. Pawson and P. Nash, Science, 2003, 300, 445–452 CrossRef CAS.
- S. Miyamoto, S. K. Akiyama and K. M. Yamada, Science, 1995, 267, 883–885 CrossRef CAS.
- R. J. Conrado, J. D. Varner and M. P. DeLisa, Curr. Opin. Biotechnol., 2008, 19, 492–499 CrossRef CAS.
- A. Fegan, B. White, J. C. T. Carlson and C. R. Wagner, Chem. Rev., 2010, 110, 3315–3336 CrossRef CAS.
- P. Ringler and G. E. Schulz, Science, 2003, 302, 106–109 CrossRef CAS.
- M. Ma and D. Bong, Org. Biomol. Chem., 2011, 9, 7296–7299 CAS.
- S. Burazerovic, J. Gradinaru, J. Pierron and T. R. Ward, Angew. Chem., Int. Ed., 2007, 46, 5510–5514 CrossRef CAS.
- R. Kluger and J. Zhang, J. Am. Chem. Soc., 2003, 125, 6070–6071 CrossRef CAS.
- M. Breurken, E. H. M. Lempens, R. P. Temming, B. A. Helms, E. W. Meijer and M. Merkx, Bioorg. Med. Chem., 2011, 19, 1062–1071 CrossRef CAS.
- V. M. Hernández-Rocamora, S. W. A. Reulen, B. de Waal, E. W. Meijer, J. M. Sanz and M. Merkx, Chem. Commun., 2011, 47, 5997–5999 RSC.
- C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 1200–1216 CrossRef CAS.
- C. J. Delebecque, A. B. Lindner, P. A. Silver and F. A. Aldaye, Science, 2011, 333, 470–474 CrossRef CAS.
- R. J. Conrado, G. C. Wu, J. T. Boock, H. Xu, S. Y. Chen, T. Lebar, J. Turnšek, N. Tomšič, M. Avbelj, R. Gaber, T. Koprivnjak, J. Mori, V. Glavnik, I. Vovk, M. Benčina, V. Hodnik, G. Anderluh, J. E. Dueber, R. Jerala and M. P. DeLisa, Nucleic Acids Res., 2011, 40, 1879–1889 CrossRef.
- B. R. Griffith, B. L. Allen, A. C. Rapraeger and L. L. Kiessling, J. Am. Chem. Soc., 2004, 126, 1608–1609 CrossRef CAS.
- S. Le Gac, E. Schwartz, M. Koepf, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem.–Eur. J., 2010, 16, 6176–6186 CrossRef CAS.
- C. W. Cairo, J. E. Gestwicki, M. Kanai and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 1615–1619 CrossRef CAS.
- O. I. Wilner, Y. Weizmann, R. Gill, O. Lioubashevski, R. Freeman and I. Willner, Nat. Nanotechnol., 2009, 4, 249–254 CrossRef CAS.
- B. A. Grzybowski, C. E. Wilmer, J. Kim, K. P. Browne and K. J. M. Bishop, Soft Matter, 2009, 5, 1110–1128 RSC.
- J.-M. Lehn, Top. Curr. Chem., 2012, 322, 1–32 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- S. Tamaru, M. Ikeda, Y. Shimidzu, S. Matsumoto, S. Takeuchi and I. Hamachi, Nat. Commun., 2010, 1, 20 CrossRef.
- D. A. Uhlenheuer, K. Petkau and L. Brunsveld, Chem. Soc. Rev., 2010, 39, 2817–2826 RSC.
- S. I. Stupp, Nano Lett., 2010, 10, 4783–4786 CrossRef CAS.
- K. Petkau-Milroy and L. Brunsveld, Org. Biomol. Chem., 2013, 11, 219–232 CAS.
- A. Barnard and D. K. Smith, Angew. Chem., Int. Ed., 2012, 51, 6572–6581 CrossRef CAS.
- L. Brunsveld, B. G. G. Lohmeijer, J. A. J. M. Vekemans and E. W. Meijer, Chem. Commun., 2000, 2305–2306 RSC.
- M. K. Müller and L. Brunsveld, Angew. Chem., Int. Ed., 2009, 48, 2921–2924 CrossRef.
- M. K. Müller, K. Petkau and L. Brunsveld, Chem. Commun., 2011, 47, 310–312 RSC.
- L. Ghosez, B. Haveaux and H. G. Viehe, Angew. Chem., Int. Ed. Engl., 1969, 8, 454–455 CrossRef CAS.
- G. Lemercier, S. Gendreizig, M. Kindermann and K. Johnsson, Angew. Chem., Int. Ed., 2007, 46, 4281–4284 CrossRef CAS.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2002, 21, 86–89 CrossRef.
- A. Juillerat, T. Gronemeyer, A. Keppler, S. Gendreizig, H. Pick, H. Vogel and K. Johnsson, Chem. Biol., 2003, 10, 313–317 CrossRef CAS.
- D. Srikun, A. E. Albers, C. I. Nam, A. T. Iavarone and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 4455–4465 CrossRef CAS.
- D. A. Uhlenheuer, D. Wasserberg, C. Haase, H. D. Nguyen, J. H. Schenkel, J. Huskens, B. J. Ravoo, P. Jonkheijm and L. Brunsveld, Chem.–Eur. J., 2012, 18, 6788–6794 CAS.
- Under SDS-PAGE and ESI-MS conditions the supramolecular polymer disassembles into monomers and only the monomeric protein–discotic conjugates can be observed.
- K. Petkau-Milroy and L. Brunsveld, Eur. J. Org. Chem., 2013 DOI:10.1002/ejoc.201300057.
- K. Petkau-Milroy, M. H. Sonntag, A. H. A. M. van Onzen and L. Brunsveld, J. Am. Chem. Soc., 2012, 134, 8086–8089 CrossRef CAS.
- S. W. A. Reulen and M. Merkx, Bioconjugate Chem., 2010, 21, 860–866 CrossRef CAS.
- R. J. Brea, M. E. Vázquez, M. Mosquera, L. Castedo and J. R. Granja, J. Am. Chem. Soc., 2007, 129, 1653–1657 CrossRef CAS.
- P. Besenius, Y. Goedegebure, M. Driesse, M. Koay, P. H. H. Bomans, A. R. A. Palmans, P. Y. W. Dankers and E. W. Meijer, Soft Matter, 2011, 7, 7980–7983 RSC.
- D. J. Toft, T. J. Moyer, S. M. Standley, Y. Ruff, A. Ugolkov, S. I. Stupp and V. L. Cryns, ACS Nano, 2012, 6, 7956–7965 CrossRef CAS.
- J. Z. Gasiorowski and J. H. Collier, Biomacromolecules, 2011, 12, 3549–3558 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, methods, detailed experimental procedures, compound and self-assembly characterization, and supplementary figures. See DOI: 10.1039/c3sc50891a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2013 |