 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWithanolide A: synthesis and structural requirements for neurite outgrowth†
Raphael Liffert‡a, Johannes Hoecker‡a, Chandan K. Jana§a, Tom M. Woods¶a, Patrick Burcha, Henning J. Jessen||a, Markus Neuburgerb and Karl Gademann*a
aDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: karl.gademann@unibas.ch; Fax: +41 612-671-105
bLaboratory for Chemical Crystallography, University of Basel, Spitalstrasse 51, 4056 Basel, Switzerland
First published on 23rd April 2013
Abstract
The exploration of different routes leading to the stereoselective synthesis of the neuritogenic steroid withanolide A is reported in full detail. Notable features of the synthesis include a Corey–Seebach homologation, a vinylogous aldol reaction, a singlet oxygen Schenck-ene reaction, and finally, a late-stage Wharton transposition. Semi-synthetic elaboration of the natural product resulted in 15 derivatives allowing a better understanding of its inherent reactivity pattern as well as its biological properties related to neuronal differentiation and neurite outgrowth.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by amyloid plaque formation, dysfunctional neurons and loss of cognitive functions.1a The number of people suffering from this disease is increasing at an alarming rate, with death occurring approximately 9 years after diagnosis.2 The pathogenesis of AD is yet to be fully understood, and both genetic and environmental factors have been suggested to contribute to its progress.3 Currently, dementia is treated mainly with acetylcholine esterase inhibitors that delay the progression rather than actually restore brain function. Due to the prevalence of AD and the nonexistence of long-term therapies, there is a strong requirement to find new drug leads for the therapy of AD and associated diseases.1bThe roots of Withania somnifera, also known as ashwagandha (Indian ginseng), are utilized in traditional ayurvedic medicine to improve the cognitive performance of the elderly.4 In addition, they have been frequently used in the context of anti-aging, memory reconstruction4 and more recently as cognitive enhancers5 for healthy subjects. Withanolide A (1) isolated from the roots of Withania somnifera,6 constitutes one of the most active compounds in the methanolic extracts of ashwagandha and has been demonstrated to possess strong pharmacological properties with regard to axonal outgrowth, regeneration of neurites and recovery of damaged synapses in mice.4,7 This pharmacological evidence encouraged us to further investigate this natural product beyond the recently completed first total synthesis of the steroid 1,8 and to prepare derivatives for structure activity relationship (SAR) studies.
Withanolide A is a highly oxygenated steroidal lactone offering several challenges in the context of its synthesis (Fig. 1). The natural product family of the withanolides is characterized by an A, B, C, D steroid ring system and a lactone side chain (SC).9,10 Withanolide A possesses a reactive enone moiety in the A ring, an epoxy alcohol in the B ring, as well as a tetrasubstituted unsaturated lactone as a side chain. The main synthetic challenges arise from the stereoselective construction of the side chain and the oxidation pattern of the A and B ring and the diastereoselective instalment of a tertiary alcohol at C-20. Departing from a cheap and commercially available starting material, we chose pregnenolone 2 for our synthetic endeavours.
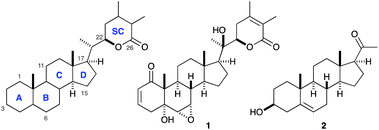 | ||
| Fig. 1 Structures of withanolide A (1) and pregnenolone (2). | ||
In this edge article, we report in full detail on different synthetic approaches to the target withanolide A culminating in the first synthesis of this steroid. In addition, semi-synthesis studies not only allowed the reactivity of the different parts of the steroidal structure to be assessed, but also enabled the generation of 15 derivatives resulting in detailed SAR studies on neuronal differentiation and neurite outgrowth in human neuroblastoma cells.
Results and discussion
Evaluation of different routes to withanolide A
We initially identified the presence of the α,β-unsaturated lactone in the side chain as the most labile fragment of withanolide A (1), which led us to embark on a strategy involving late instalment of this pyranone. It was further anticipated that we could use a procedure developed by Ikekawa and co-workers11 to form the lactone side chain motif from a simple steroidal aldehyde precursor by a vinylogous Mukaiyama aldol reaction in the last step, after the B and A rings had been completed.The approach started with the known homologation required for the side chain by a Corey–Seebach umpolung reaction.12 Therefore, the 3β-OH group of commercially available pregnenolone (2) was protected as its TBS ether and treatment of the resulting ketone with lithiated dithiane afforded the tertiary alcohol 3 (Scheme 1) in 95% yield (dr 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Several methods were investigated to cleave the dithiane moiety in 3, with the main concern being the stability of the TBS protecting group. Methods such as HgCl2, NBS and I2 both with and without addition of base were largely unsuccessful and resulted in either the loss of the TBS group as well as the dithioacetal or more complex mixtures. However, treatment of dithiane 3 with NCS in a CH2Cl2–H2O mixture in an open vessel at room temperature afforded the desired α-hydroxy aldehyde in a good yield. Protection of the C-20 tertiary alcohol as its MOM ether 4 was achieved by treating a solution of the alcohol and N,N-diisopropylethylamine (DIPEA) with MOMI and refluxing the reaction overnight (in 69% over 2 steps). The efficiency of the reaction was found to be dependent on the quality of the starting materials, in particular, NaI was dried under high vacuum at 100 °C for several hours and MOMCl was freshly distilled before use. A simple filtration through a short pad of silica was sufficient to provide material for the next step. Reduction of aldehyde 4 with NaBH4 in MeOH proceeded uneventfully and furnished the desired alcohol as a white solid, which was used directly in the next step. Treatment of the alcohol with NaH followed by addition of PMBCl and a catalytic amount of tetrabutylammonium iodide (TBAI) afforded the desired differentially protected triol 5.
:1). Several methods were investigated to cleave the dithiane moiety in 3, with the main concern being the stability of the TBS protecting group. Methods such as HgCl2, NBS and I2 both with and without addition of base were largely unsuccessful and resulted in either the loss of the TBS group as well as the dithioacetal or more complex mixtures. However, treatment of dithiane 3 with NCS in a CH2Cl2–H2O mixture in an open vessel at room temperature afforded the desired α-hydroxy aldehyde in a good yield. Protection of the C-20 tertiary alcohol as its MOM ether 4 was achieved by treating a solution of the alcohol and N,N-diisopropylethylamine (DIPEA) with MOMI and refluxing the reaction overnight (in 69% over 2 steps). The efficiency of the reaction was found to be dependent on the quality of the starting materials, in particular, NaI was dried under high vacuum at 100 °C for several hours and MOMCl was freshly distilled before use. A simple filtration through a short pad of silica was sufficient to provide material for the next step. Reduction of aldehyde 4 with NaBH4 in MeOH proceeded uneventfully and furnished the desired alcohol as a white solid, which was used directly in the next step. Treatment of the alcohol with NaH followed by addition of PMBCl and a catalytic amount of tetrabutylammonium iodide (TBAI) afforded the desired differentially protected triol 5.
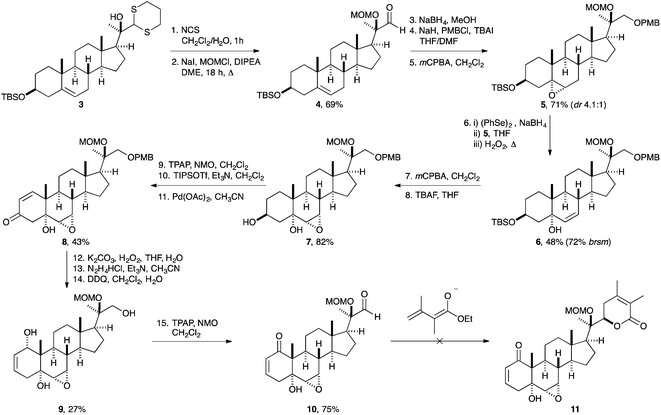 | ||
| Scheme 1 Approach towards the synthesis of MOM-protected withanolide A derivative 11. | ||
With the homologated triol 5 in hand the stage was set to target the functionalization on the B ring. Epoxidation of the double bond proceeded smoothly affording a 4.1:1 diastereomeric ratio of the desired α-epoxide 5 in 71% yield over three steps. Performing the reaction at 0 °C slightly improved the selectivity, but lower temperatures (e.g. −15 °C) reduced the rate of reaction. Following a method of Sharpless and Lauer for the oxidative opening of epoxides,13 reduction of (PhSe)2 with NaBH4 in EtOH followed by addition to the MOM protected epoxide and further treatment with H2O2, afforded the desired allylic alcohol 6 in 48% yield accompanied by recovered starting material. The undesired β-epoxide was not reactive towards these conditions and separation was easily achieved. Allylic alcohol 6 was sensitive to acidic conditions and was prone to rearrange to the thermodynamically more stable trisubstituted olefin. The allylic alcohol 6 was then further transformed to the epoxy alcohol by simple treatment with mCPBA in CH2Cl2. This directed epoxidation (by the tertiary OH group) was found to be completely selective and high yielding and completed the functionalization of the B-ring. Deprotection of the TBS group by treatment with tetrabutylammonium fluoride (TBAF) furnished compound 7 bearing a free OH group at C3.
For the subsequent transformation of the A ring to the desired enone, allylic oxidation reactions turned out to be unsuccessful,14 and therefore Saegusa oxidation was chosen instead.15 After oxidation of the secondary OH group by TPAP, the formed TIPS enol ether was treated with 1 equivalent of Pd(OAc)2 in CH3CN and the desired α,β-unsaturated ketone 8 was isolated in 43% yield (over three steps). Attempts to reduce the amount of Pd(OAc)2 employed by adding benzoquinone as a co-oxidant led to poor yields of the desired product. Epoxidation of the unsaturated ketone under basic conditions gave the bis-epoxide in 54% yield with complete diastereoselectivity. K2CO3 was found to be the best choice of base; stronger bases such as NaOH and KOtBu gave complex mixtures of products. With the keto epoxide in hand the stage was set for the Wharton transposition. Wharton's original conditions (N2H4·H2O, AcOH) afforded poor yield of the desired allylic alcohol.16 However, in 1989 Dupuy and Luche reported modified conditions for the Wharton transposition using basic conditions,17 and in such a way, the conversion to the allylic alcohol was improved. Subsequent deprotection of the PMB protecting group using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) proceeded smoothly and afforded triol 9 in a 50% yield over the two steps. The primary and secondary diols were then oxidized simultaneously with TPAP, NMO to complete the A ring functionalities and to furnish the desired C-22 aldehyde 10. In order to complete the synthesis of withanolide A (1), the side chain lactonization was evaluated next. However, despite using a variety of conditions, the final lactonization to 11 could not be performed successfully. It is likely that the presence of the enone in the A ring is detrimental to this approach, which therefore had to be abandoned.
Preparation of withanolide A
Having realized the sensitivity of the A ring, both from the unsuccessful synthetic attempt as discussed above, and from the ease of its functionalization in the context of semi-synthesis studies (vide infra), we sought to introduce this enone last in the synthetic sequence. In addition, these studies hinted at the stability of the side chain lactone, which led us to choose its early instalment (Scheme 2). Therefore, following the procedure developed by Ikekawa and co-workers,10a,11 the aldehyde 4 was reacted with the vinylogous enolate from ethyl 2,3-dimethylbut-2-enoate and LiHMDS in a mixture of THF and DMPU via a stereoselective aldol reaction to deliver the unsaturated lactone side chain 12 in very good yield and stereoselectivity (87%, dr 93:7). Again, protection of the tertiary alcohol as the MOM ether was found to be key for this transformation. Unselective side-reactions during exposure to strong bases during its introduction were avoided, and the relatively small size of the MOM group meant that the unhindered nucleophilic attack of the formed enolate on the aldehyde was possible.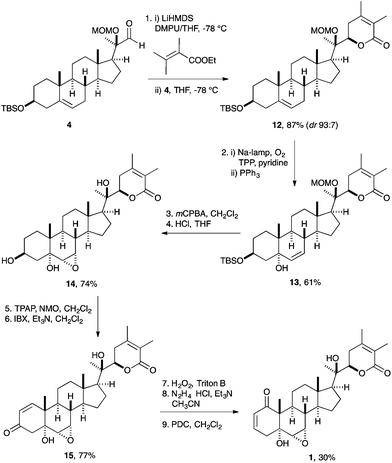 | ||
| Scheme 2 Synthesis of withanolide A (1) from aldehyde 4. | ||
We next proceeded to install the correct oxidation pattern in the B ring of withanolide A. The challenge as previously described consisted in the regioselective instalment of the epoxy alcohol in the presence of the unsaturated lactone. Having struggled with limitations associated with phenylselenium-chemistry as detailed above, we recognized the singlet oxygen mediated photo-oxygenative olefin migration (Schenck-ene reaction18) as a straightforward method19 for the preparation of the allylic alcohol 13 from an unfunctionalized olefin. Therefore, olefin 12 was allowed to react with singlet oxygen generated in situ from O2 in the presence of meso-tetraphenylporphyrin (TPP) as sensitizer under irradiation with Na-light. The formed hydroperoxide was in situ reduced by PPh3 to the allylic alcohol 13 in good yield (61%). Directed epoxidation of allylic alcohol 13 mediated by mCPBA proceeded as anticipated to provide, after removal of all protecting groups by exposure to HCl, the desired epoxy alcohol 14 in excellent yield.
It should be pointed out that we have, by design, chosen to avoid any protecting groups for the remaining elaboration of triol 14 therefore exploiting the inherent reactivity of this steroid derivative. The final installation of the correct oxidation pattern in the A ring started by oxidation of the triol 14 first by TPAP and NMO and second by exposure to 2-iodoxybenzoic acid (IBX)20 to provide the corresponding enone 15 (77%). The unsaturated ketone 15 was then converted with basic aqueous H2O2 (Triton B)21 to the epoxy ketone. The stage was again set for the Wharton transposition on the epoxy ketone substrate. Therefore, the intermediate was subjected to reaction with hydrazine (N2H4·HCl) in the presence of base to furnish the rearranged allylic alcohol.17 This reaction proceeded smoothly to give, after subsequent oxidation by pyridinium dichromate (PDC), withanolide A (1) in good yield (30% over three steps).
All analytical data (1H and 13C NMR, optical rotation, UV, m.p.) for the synthetic and isolated natural product were found to be in full agreement. The final proof of identity of the synthetic and isolated natural sample of 1 was established by co-injection and analysis of natural and synthetic samples by HPLC.
Semi-synthesis of new derivatives of withanolide A
In order to access additional derivatives for biological studies, we initiated semi-synthesis studies on withanolide A (1). This steroidal lactone was therefore isolated from the dried and powdered roots of Withania somnifera (ashwagandha) by a modification of a known procedure.6 After an extensive survey of different approaches commonly employed in semi-synthesis to functionalize withanolide A (1), we found that only the A ring was susceptible to chemical modification. In addition, A ring derivatives bearing e.g. a 3β-hydroxy group like sominone—an aglycon of withanoside IV—were found to display neuritogenic properties as shown from the work of Tohda and co-workers,22 lending further support to the hypothesis that compounds with variations in this ring may retain biological activity.We first targeted the regioselective 1,2-reduction of withanolide A resulting in the allylic alcohol 16, which was envisioned to serve as a building block for a variety of new functionalities at C-1. The reduction was performed under Luche conditions,23 using CeCl3 and NaBH4 in a mixture of MeOH and CHCl3 in 75% yield with complete diastereoselectivity (Scheme 3). Surprisingly, the same stereochemical outcome was observed when using simple NaBH4 without additional CeCl3 to coordinate the ketone function, as established by an X-ray crystal structure analysis in an earlier study.8 Unfortunately, all attempts to further functionalize the C-1 hydroxy group or acetylation reactions were unsuccessful. Regioselective hydrogenation of the 2,3-double bond was performed by using 2 mol% Crabtree's catalyst under a hydrogen atmosphere of 10 bar to obtain the saturated alcohol 17.
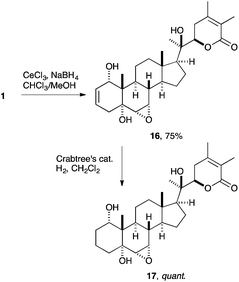 | ||
| Scheme 3 Regioselective reduction of withanolide A. | ||
Due to the problems associated with the manipulation of the allylic alcohol 16, a new strategy for the derivatization of the natural product needed to be developed. Tohda et al. showed in their studies on sominone that steroids bearing a C3-OH group can serve as useful intermediates.22 Therefore we targeted a synthetic route from withanolide A to C3-OH derivatives, as shown in Scheme 4. The synthetic plan relied on epoxidation of withanolide A (1) followed by epoxide opening under reductive conditions, which was used in the context of B ring functionalization (see 5 to 6, Scheme 1). It should be pointed out that again no protecting groups were introduced and that these reagents have been found to be compatible with the functional group manifold present in the A and B rings and the side chain lactone.
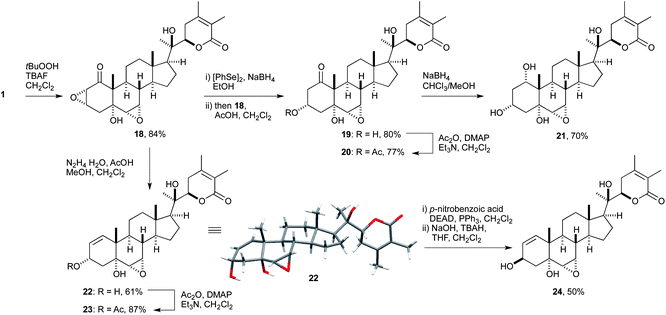 | ||
| Scheme 4 Semi-synthetic transformations of withanolide A to access A ring derivatives without the enone moiety. | ||
We first evaluated the epoxidation of withanolide A (1) by using the conditions developed in the course of the total synthesis. Exposure of steroid lactone 1 to aqueous H2O2 in the presence of Triton B afforded epoxide 18 in 48% yield on small scale. Unfortunately, the reaction was dependent on the scale, and the use of 100 mg of 1 resulted in a dramatic loss of material (11% yield). After extensive investigation of different conditions (solvent, base and oxidizing reagents)24 we found the fluoride-promoted epoxidation of α,β-unsaturated carbonyl compounds described by Yoshikoshi and co-workers was compatible with our system.25 By using their protocol, the use of TBAF as base and H2O2 or tert-butyl hydroperoxide (tBuOOH) as oxidation reagent was found to be superior. These conditions improved the yield reliably to 84% (on a 150 mg scale), with CH2Cl2 as solvent and tBuOOH as oxidant.
The organoselenium-mediated reduction of α,β-epoxy ketone 18 to the corresponding β-hydroxy ketone was investigated next. Alcohol 19 was obtained in good yields on adding an ethanolic solution of the active reagent (Na[PhSeB(OEt)3]) and AcOH to a solution of epoxy ketone 18 in CH2Cl2.26 The chemical shift of the equatorial C3-H in 19 was found to be at 4.11 ppm, whereas the axial proton reported for the C3 diastereomeric natural product27 resonates at 4.91 ppm. This large difference might be caused by the influence of the neighbouring hydroxyl group on the axial proton and could be observed later for the allylic alcohols 22 and 24 as well. Efficient acetylation of alcohol 19 under standard conditions (Ac2O, DMAP, Et3N in CH2Cl2) delivered acetate 20 in 77% yield without any detectable amount of eliminated side-product. This example demonstrates the feasibility of introducing carboxylate esters of choice at this position. Reduction of ketone 19 was performed using NaBH4 and afforded a new baseline spot by TLC. UPLC-MS confirmed formation of tetraol 21, but the isolation of this hydrophilic compound turned out to be difficult.28 Finally, work-up with a saturated solution of Rochelle salt liberated tetraol 21 and allowed the characterization of its structure by NMR spectroscopy. The stereochemical outcome of 21 was anticipated from the NaBH4 mediated reduction of withanolide A (1) and could be confirmed by NOESY NMR spectroscopy. The structural assignment was supported by NOE cross peaks of H-1 with H-19 and H-2 and H-4, respectively.
Concerning the targeted introduction of substituents at C3, another possibility could be the transformation of epoxy ketone 18 to allylic alcohol 22 (Scheme 4). For this manipulation, the Wharton carbonyl transposition was investigated using conditions as discussed earlier. Consequently, epoxy ketone 18 was treated with NH2NH2·HCl in the presence of Et3N in CH3CN,16 but no product formation was observed. Typical acidic conditions with NH2NH2·H2O and AcOH for Wharton transposition are reported in various publications and O'Doherty and Wang described a method using more equivalents of both reagents NH2NH2·H2O and AcOH to improve the yield.29 They also noted that a protic solvent can be favourable for certain substrates. Due to the low solubility of 18 in methanol, the reaction was successfully conducted in a 1:1 mixture of methanol and CH2Cl2 to obtain the allylic alcohol 22 in 61% yield after 3 h at room temperature. The structure of alcohol 22 was confirmed by X-ray analysis of the crystal structure and the configuration of the 3α-OH group was retained from the epoxidation step.
Next, we investigated the introduction of functional groups at the newly formed secondary alcohol in order to probe if alcohol 22 can be attached to various residues such as, for example, sugars known from the studies on sominone by Tohda and co-workers. Acetylation of allylic alcohol 22 was performed again under standard acetylation conditions and afforded acetate 23 in good yield. This indicates that alcohol 22 is also a feasible precursor for further manipulations. Inversion of the C3 hydroxyl group in 22 was accomplished using the Mitsunobu procedure. Esterification with para-nitrobenzoic acid, followed by saponification with aqueous NaOH in the presence of tetrabutylammonium hydroxide (TBAH) as phase-transfer catalyst afforded 3β-allylic alcohol 24 in 50% yield.
Additionally, the double bond of allylic alcohol 22 was reduced with 10 mol% Pd on activated charcoal. Quantitative conversion provided alcohol 25 as an inseparable 1:1 mixture with the isomerized saturated ketone 14a (Scheme 5), an intermediate obtained in our synthesis of 1 (see Scheme 2).30 Given that we were interested in alcohol 25, Crabtree's catalyst turned out to be efficient for the hydrogenation of 22 to yield alcohol 25 quantitatively without the isomerized product.
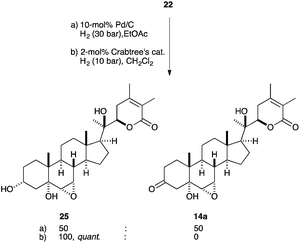 | ||
| Scheme 5 Hydrogenation of allylic alcohol 22. | ||
Oximes of withanolide A as prodrugs may improve the solubility as well as the bioavailability in vivo. Additionally, oximes were found to be the only possibility to modify the C1 position of natural product 1 (Scheme 6). The oximes 26–30 were easily obtained by heating withanolide A 1 or the derived epoxide 18 and the corresponding hydroxylamine hydrochloride in pyridine at 70 °C for 1 to 2 days in good yields.
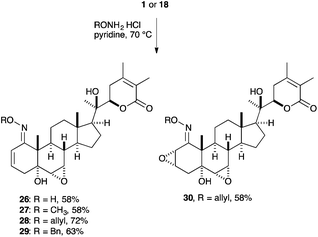 | ||
| Scheme 6 Oxime formation of withanolide A. | ||
Biological evaluation
The natural product 1 and several semi-synthetic derivatives were tested in order to determine their neuritogenic properties according to the previously published methods.7a,8 Human SH-SY5Y neuroblastoma cells (4 × 104 cells per cm2) were grown on collagen I coated 24-well plates in minimal essential medium (MEM) with 5% fetal bovine serum (FBS). After incubation for 4 days in the presence of the corresponding compounds (1 μM, 0.1% DMSO), the cells were examined under a phase contrast microscope after fixation (5% formaldehyde in PBS buffer solution), stained (modified Giemsa stain), and the differentiated cells were counted. For each compound nine pictures from random areas of three different wells were taken (for representative micrographs see ESI†). At least 500 cells were counted for each individual compound. The criterion for a differentiated cell was at least one neurite with a length of more than 50 μm. In control experiments DMSO (0.1%) was used as a negative control. The results are summarized in Fig. 2.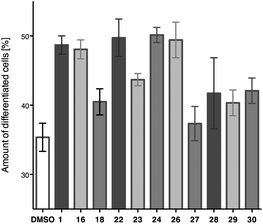 | ||
| Fig. 2 Neurite outgrowth activity of DMSO blank, withanolide A and derivatives in human SH-SY5Y cells with MEM as cell-medium. Error bars denote SEM. | ||
While it is difficult to draw quantitative conclusions from the biological assays, several facts became apparent: compounds 16, 22, 24, and 26 showed similar activities when compared to withanolide A with regard to neurite outgrowth stimulating activity in SH-SY5Y cells. The compounds 18, 23, and 27–30 showed in fact less activity than withanolide A, to the extent that some compounds can be hardly distinguished from the solvent control in these phenotypic assays. It appears that there is a correlation between the size of the attached group and activity. For example, the hydroxylamine 26 shows increased activity than the corresponding and sterically more demanding oximes 27–30. The same trend was observed for allyl alcohol 22 and its acetate 23. Additionally, the chemically reactive enone moiety of withanolide A in the A ring might not be required for neuroactive properties, as the allylic alcohols 16 and 22 retained activity.
Conclusions
In conclusion, we have developed the first successful synthesis of the pharmacologically important steroid withanolide A by adjusting the route based on the understanding of the inherent reactivity of the natural product by semi-synthesis studies. Notable features of this synthesis include (1) a highly diastereoselective Schenck-ene reaction, (2) a minimal protecting group strategy exploiting the inherent reactivity pattern in the endgame and (3) a Wharton transposition for the A ring formation. Further, we have synthesised more than 15 derivatives of withanolide A directly from the isolated natural product. Therefore, we developed two new synthetic routes that allow the generation of a hydroxyl group at C3 and thereby open the door for attachment of a variety of functionalities. Both key intermediates, the β-hydroxy ketone 19 and the allylic alcohol 22, were synthesised in high yields and full diastereoselectivity using simple reaction conditions in two steps from 1.In addition, biological studies of neurite outgrowth in human SH-SY5Y cells demonstrated the dependence of neuritogenic properties on the size rather than the functionality in the A ring of the evaluated compounds. Three compounds indicated a comparable or higher neurite outgrowth activity in cell assays than withanolide A itself. Further investigations towards the modification of functional groups at C-3 of withanolide A would probably give access to more sophisticated candidates and allow for the manipulation of properties such as solubility or bioavailability.
Acknowledgements
Financial support by the Swiss National Science Foundation (200021-144028 and PE002-117136/1) and DFG (Project JE 572/1-1) is gratefully acknowledged. We thank Novartis for supporting part of this work (Novartis Early Career Award 2010 to K.G.). We thank Dr. Hideki Miyatake Ondozabal for proof-reading of this manuscript.Notes and references
-
(a) Alzheimer's Disease International, World Alzheimer Report, 2012, pp. 1–13;
(b) for selected recent studies on neuritogenic compounds, see, for example: S. P. Waters, Y. Tian, Y.-M. Li and S. J. Danishefsky, J. Am. Chem. Soc., 2005, 127, 13514–13515 CrossRef CAS
; R. M. Wilson and S. J. Danishefsky, Acc. Chem. Res., 2006, 39, 539–549 Search PubMed
- For a review, see: P. Williams, A. Sorribas and M.-J. R. Howes, Nat. Prod. Rep., 2011, 28, 48–77 RSC
- D. K. Lahiri, B. Maloney, M. R. Basha, Y. W. Ge and N. H. Zawia, Curr. Alzheimer Res., 2007, 4, 219–228 CrossRef CAS
- For a review on the neuroactive properties of ashwagandha, see: S. Jain, S. D. Shukla, K. Sharma and M. Bathnagar, Phytother. Res., 2001, 15, 544–548 CrossRef CAS
- http://www.cerebralhealth.com/brainhealthsupplements.php.
- S. S. Subramanian, P. D. Sethi, E. Glotter, I. Kirson and D. Lavie, Phytochemistry, 1971, 10, 685–688 CrossRef CAS
- For neuroactive properties of withanolide A, see:
(a) T. Kuboyama, C. Tohda, J. Zhao, N. Nakamura, M. Hattori and K. Komatsu, NeuroReport, 2002, 13, 1715–1720 CrossRef CAS
- C. K. Jana, J. Hoecker, T. Woods, H. J. Jessen, M. Neuburger and K. Gademann, Angew. Chem., Int. Ed., 2011, 50, 8407–8411 CrossRef CAS
- For reviews on withanolide structures, bioactivities and synthetic approaches, see:
(a) L.-X. Chen, H. He and F. Qiu, Nat. Prod. Rep., 2011, 28, 705–740 RSC
- For synthetic investigations on withanolides, see:
(a) K. Gamoh, M. Hirayama and N. Ikekawa, J. Chem. Soc., Perkin Trans. 1, 1984, 449–454 RSC
- M. Ishiguro, M. Hirayama, H. Saito, A. Kajikawa and N. Ikekawa, Heterocycles, 1981, 15, 823–834 CrossRef CAS
- B. B. Shingate, B. G. Hazra, V. S. Pore, R. G. Gonnade and M. Bhadbhade, Tetrahedron, 2007, 63, 5622–5635 CrossRef CAS
- K. B. Sharpless and R. F. Lauer, J. Am. Chem. Soc., 1973, 95, 2697–2699 CrossRef CAS
-
(a) J.-Q. Yu, H.-C. Wu and E. J. Corey, Org. Lett., 2005, 7, 1415–1417 CrossRef CAS
- Y. Ito, T. Hirao and T. Saegusa, J. Org. Chem., 1978, 43, 1011–1013 CrossRef CAS
- P. S. Wharton and D. H. Bohlen, J. Org. Chem., 1961, 26, 3615–3616 CrossRef CAS
- C. Dupuy and J. L. Luche, Tetrahedron, 1989, 45, 3437–3444 CrossRef CAS
-
(a) G. O. Schenck, DE-B 933925, 1943
-
(a) W. Adam and E. Staab, Liebigs Ann. Chem., 1988, 757–759 CrossRef CAS
- K. C. Nicolaou, T. Montagnon and P. S. Baran, Angew. Chem., Int. Ed., 2002, 41, 993–996 CrossRef CAS
- M. T. Barros, C. D. Maycock and M. R. Ventura, Tetrahedron, 1999, 55, 3233–3244 CrossRef CAS
-
(a) T. Kuboyama, C. Tohda and K. Komatsu, Eur. J. Neurosci., 2006, 23, 1417–1426 CrossRef
- A. L. Gemal and J. L. Luche, J. Am. Chem. Soc., 1981, 103, 5454–5459 CrossRef CAS
-
(a) F. E. Ziegler, K.-J. Hwang, J. F. Kadow, S. I. Klein, U. K. Pati and T.-F. Wang, J. Org. Chem., 1986, 51, 4573–4579 CrossRef CAS
- M. Miyashita, T. Suzuki and A. Yoshikoshi, Chem. Lett., 1987, 285–288 CrossRef CAS
-
(a) M. Miyashita, T. Suzuki and A. Yoshikoshi, Tetrahedron Lett., 1987, 28, 4293–4296 CrossRef CAS
- M. I. Choudhary, S. Yousuf, S. A. Nawaz, S. Ahmed and A. Rahman, Chem. Pharm. Bull., 2007, 52, 1358–1361 CrossRef
- Baran and co-workers reported that similar 1,3,5-trihydroxy cyclohexane steroids showed a tenacity for glass: H. Renata, Q. Zhou and P. Baran, Science, 2013, 339, 59–63 CrossRef CAS
- H. Y. Wang and G. A. O'Doherty, Chem. Commun., 2011, 47, 10251–10253 RSC
- R. Uma, C. Crévisy and R. Grée, Chem. Rev., 2003, 103, 27–52 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, crystal data and CIF file for alcohol 22. CCDC 924130. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc50653c |
| ‡ Both authors contributed equally to this work. |
| § Present address: Department of Chemistry, Indian Institute of Technology Guwahati, 781039 Guwahati, Assam, India. |
| ¶ Present address: Department of Chemistry, The University of Auckland, 23 Symonds Street, Auckland CBD, New Zealand. |
| || Present address: Institute of Organic Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland. |
| This journal is © The Royal Society of Chemistry 2013 |