 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An enantioselective tandem reduction/nitro-Mannich reaction of nitroalkenes using a simple thiourea organocatalyst†
James C.
Anderson
* and
Paul J.
Koovits
Department of Chemistry, University College London, 20 Gordon Street, WC1H 0AJ, UK. E-mail: j.c.anderson@ucl.ac.uk; Tel: +44 (0)20 7679 5585
First published on 2nd May 2013
Abstract
There is a continual need for ever more effective and operationally simpler methods for the asymmetric synthesis of nitrogen containing molecules. We report here a generally efficient synthesis of stereochemically defined β-nitroamine building blocks which, through the combination of two catalytic transformations into one tandem process, results in the use of a simpler asymmetric catalyst, less reaction materials, shorter reaction times, circumvents the need for moisture sensitive reaction partners and leads to a wider substrate scope. Using para-methoxy-phenyl (PMP) protected imines, a Hantzsch ester as hydride source and a simple and economic thiourea organocatalyst, we have promoted the nitro-Mannich reaction with a nitroalkene to form anti-β-nitroamines. After protection as their trifluoroacetamides the products can be isolated in good yields (32–83%), high diastereomeric ratios (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to >95:5) and excellent enantioselectivity (73–99% ee).
:10 to >95:5) and excellent enantioselectivity (73–99% ee).
Introduction
Functional groups containing nitrogen are present in 75% of all drug molecules, in every major drug class and in a wide variety of biologically active natural products.1 The β-nitroamine functionality has emerged as a privileged building block due to its complimentary synthetic flexibility available from the two different nitrogen atom oxidation states. As such it has been applied in the synthesis of many nitrogen containing functional groups including α-amino carbonyls,2 1,2-diamines,3 peptidomimetics,4 natural products5 and many heterocyclic small molecules6 of importance to drug discovery. This stereodefined bifunctional building block is most commonly made by an enantioselective nitro-Mannich (or aza-Henry) reaction (Scheme 1). Enantioselective reactions have been controlled by asymmetric metal centered Lewis acids; chiral hydrogen bond donors, in particular by the use of asymmetric thiourea organocatalysts; chiral Brønsted acids; phase transfer catalysts and Brønsted base catalysts.7 However, many enantioselective nitro-Mannich reactions largely suffer from several limitations including a requisite for a large excess of nitroalkane and/or long reaction times;8 limited availability of complex nitroalkanes; and in the majority of cases the use of moisture sensitive N-Boc imines (Scheme 1a). The demand for making complex molecular architectures with high operational efficiencies has led to the development of many complex asymmetric catalysts.9 One way of simplifying these procedures is to combine catalytic reactions in a tandem process.10 We report a solution to these problems using an asymmetric tandem reductive nitro-Mannich strategy, with nitroalkenes and N-PMP imines, catalysed by a simple thiourea organocatalyst (Scheme 1b).11 The combination of these two catalytic transformations into one tandem process led to the use of a simpler asymmetric catalyst, less equivalents of nitroalkene and reductant, shorter reaction times and circumvented the need for moisture sensitive N-Boc imines. This concept generally may simplify other catalytic systems and specifically will allow the wider synthesis of this versatile building block.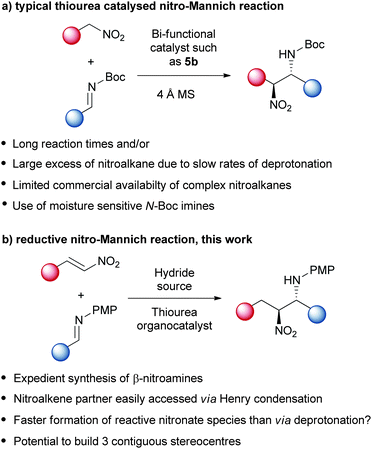 | ||
| Scheme 1 Comparison of previous work. | ||
Results and discussion
Proof of concept and reaction optimisation
Both nitro-Mannich reactions12,13 and reductions of nitroalkenes14 have been shown to be amenable to asymmetric thiourea organocatalysis, so a tandem reaction appeared plausible. Initially, we tested catalyst 5a, previously shown by Jacobsen to be effective for nitro-Mannich reactions,13b in a tandem reduction/nitro-Mannich reaction with β-nitrostyrene 1a and Hantzsch ester 4 as hydride source (Table 1). The reaction with N-Boc protected imine failed to give the desired product, instead preferentially reducing N-Boc imine (Table 1, Entry 1). However by switching to the less electrophilic N-PMP protected imine we were able to promote the desired tandem reductive nitro-Mannich reaction. Due to the instability of the β-nitroamine products, an in situ trifluoroacetamide protection was performed,15 and 3 was isolated in 29% yield and in 58% ee after 1 h (Table 1, entry 2). Extended reaction times initially gave an increase in the reaction yield, up to 9 h when β-nitrostyrene 1a had been fully consumed (Table 1, entry 3). Further ageing of the reaction however gave a decreased yield and lower enantioselectivity due to retro-addition (Table 1, entry 4). Interestingly, when using ethyl ester substituted Hantzsch ester 4b only 10% nitroalkene reduction was observed and <5% conversion to desired product 3 (Table 1, entry 5).16 Before undertaking a catalyst screen, the stoichiometry of the reaction was examined and it was discovered that by employing 2 equivalents each of the nitroalkene and hydride source the reaction was faster and higher yielding with little effect on the enantioselectivity of the reaction (Table 1, entry 6). Increasing the stoichiometry of imine had no effect on the relative rate of reaction (Table 1, entry 7) suggesting that the reduction of the nitroalkene is the rate limiting step. The reaction was also attempted in a stepwise manner where imine 2 was charged after complete reduction of 1 (Table 1, entry 8). This resulted in no formation of desired product 3 prompting us speculate that the two reactions occur in tandem.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | PG | R | Rxn time (h) | Yield (%) | dra | eeb (%) |
| a Determined by 1H NMR analysis. b Determined by chiral HPLC see ESI. c 2 equiv. of 1 and 4a. d 2 equiv. of 2. e Reaction performed using a stepwise addition. | ||||||
| 1 | Boc | CO2tBu | 16 | <5 | — | — |
| 2 | PMP | CO2tBu | 1 | 29 | 95:5 |
58 |
| 3 | PMP | CO2tBu | 9 | 45 | 95:5 |
52 |
| 4 | PMP | CO2tBu | 22 | 23 | 90:10 |
34 |
| 5 | PMP | CO2Et | 22 | <5 | — | — |
| 6c | PMP | CO2tBu | 2 | 77 | 95:5 |
50 |
| 7d | PMP | CO2tBu | 1 | 30 | 95:5 |
57 |
| 8e | PMP | CO2tBu | 9 | <5 | — | — |

Optimisation of asymmetric catalyst
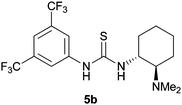
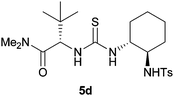
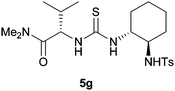
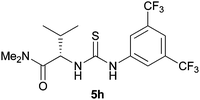
Using these optimised conditions we then screened the reaction with a variety of thiourea organocatalysts (Table 2).17 Takemoto's catalyst 5b,18 failed to reduce nitroalkene 1a and consequently no nitro-Mannich reaction was observed. Using catalyst 5c, which has been previously used for asymmetric reductions of nitroalkenes,14 we obtained a high degree of enantioselectivity (86% ee) but in a poor yield (25%, extended reaction times did not lead to an improvement in yield). Toluenesulfonamide catalyst 5d gave the desired product with excellent enantioselectivity (90% ee) and a moderate yield of 48% (extended reaction times led to a maximum yield of 75% with an 88% ee). To explore which chiral group was responsible for stereoinduction, we screened catalyst 5e, which gave the opposite enantiomer to that obtained with 5d with 64% ee; and diastereomeric catalyst 5f, which gave the same enantiomer as that obtained with 5d, albeit with a slightly reduced yield and enantioselectivity (37%, 76% ee). These two results suggest that the diaminocyclohexane group is not responsible for the high degree of stereoselectivity. Due to the relatively high cost of un-natural amino acid L-tert-leucine, we also synthesised catalyst 5g using the significantly cheaper L-valine and were surprised to observe the same high levels of enantioselectivity (90% ee). Combining these results we replaced the diaminocyclohexane moiety with an aryl group to give catalyst 5h which achieved a high level of enantioinduction (90% ee) and yield (75%). With catalyst 5h showing good potential we conducted a solvent screen that confirmed toluene as the best choice (see ESI†). Further investigation of the reaction in toluene at rt showed complete consumption of the imine after 30 min and immediate protection enabled isolation of 3 in 74% yield with an impressive 94% ee. This result suggested that stereoselectivity was probably being eroded at rt due to the reversibility of the nitro-Mannich reaction. In order to retard the retro-addition reaction we performed the reaction at lower temperatures (see ESI†). At −20 °C the desired product was isolated in excellent yield and stereoselectivity (81% yield, >95:5 dr and 98% ee) after 20 h. Prolonged reaction at −20 °C did not lead to a decrease in enantioselectivity suggesting no retro-addition occurs (see ESI†).
| a Reaction of N-PMP imine (0.14 mmol) with 1a (2 equiv.), 4a (2 equiv.) and 5 (10 mol%) in toluene (1 mL) at rt for 2 h. b Reaction performed at −20 °C over 24 h. | |
|---|---|

|
|
| 87% yield, 95:5 dr, 50% ee |
No reaction |

|
|
| 25% yield, >95:5 dr, 86% ee |
48% yield, >95:5 dr, 90% ee |

|

|
| 36% yield, >20:1 dr, −65% ee |
37% yield, 95:5 dr, 76% ee |
|
|
| 72% yield, >95:5 dr, 90% ee |
75% yield, 90:10 dr, 90% ee 81% yield, 95:5 dr, 90% eeb |
Reaction optimisation with respect to imines 2
With these optimised conditions we examined the scope of the reaction, initially with respect to imines 2 (Table 3). Both electron rich and electron poor aromatic imines (Table 3, entries 2–7) gave uniformally high yields and excellent stereoselectivities, although the ortho-trifluoromethyl substituted electron deficient imine (Table 3, entry 5) was less enantioselective (80% ee). The crude diastereoselectivities for all the ortho-substituted substrates (Table 3, entries 5–7) were lower than that of the purified products. All of the syn-diastereoisomers isolated from these analogues were of low enantiopurity which may suggest that there are different transition states in operation for each diastereoisomer. The reaction worked well for heterocyclic imines (Table 3, entries 8–9), but less so for an alkyl npentyl substituted product 3aj which was formed in moderate 73% ee (Table 3, entry 10).|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | Product | Yielda (%) | drb | eec (%) |
| a Isolated yield of 3. b dr of isolated 3 calculated by 1H NMR, parentheses indicate dr of crude product. c Determined by chiral HPLC, parenthesis indicate ee of syn-diastereomer if isolated. d Reaction performed at rt over 1 h. | |||||
| 1 | Ph | 3aa | 81 | >95:5 (>95:5) |
98 |
| 2 | p-MeOPh | 3ab | 75 | >95:5 (>95:5) |
97 |
| 3 | o-MeOPh | 3ac | 83 | >95:5 (>95:5) |
99 |
| 4 | p-F3CPh | 3ad | 74 | 90:10 (90:10) |
94 |
| 5 | o-F3CPh | 3ae | 66 | >95:5 (80:20) |
80 (4) |
| 6 | o-MePh | 3af | 75 | >95:5 (85:15) |
90 (34) |
| 7 | o-BrPh | 3ag | 70 | >95:5 (80:20) |
92 (10) |
| 8 | 2-Furyl | 3ah | 77 | 90:10 (90:10) |
97 |
| 9 | 2-Pyridyl | 3ai | 76 | >95:5 (>95:5) |
96 |
| 10d | n Pentyl | 3aj | 59 | >95:5 (>95:5) |
73 |

The yield of the reaction was also reduced due the instability of the npentyl substituted imine 2j, even at −20 °C. It was found that the yield was best at rt, where the reaction was rapid, without any noticeable effect on the enantioselectivity allowing isolation of 3aj in 59% yield.
Reaction optimisation with respect to nitroalkenes 1
The nitroalkene partner tolerated alkyl substituents, substituted aromatics and heterocyclic analogues exceptionally well giving high yields and excellent stereoselectivity (Table 4, entries 1–7). A decrease in the rate of the reaction was observed when using more electron rich nitroalkenes (Table 4, entries 6–8). This effect was most evident when using 2-furyl nitroalkene 1i which required a reaction time of 10 days to consume all of imine 2a (Table 4, entry 8). This increased reaction time also led to decreased yields due to competitive reduction of imine 2. This result also suggests that the reduction is the rate determining step of the reaction.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | Product | Rxn time (h) | Yielda (%) | drb | eec (%) |
| a Isolated yield of 3. b dr of crude 3 calculated by 1H NMR, which remained unchanged after purification. c Determined by chiral HPLC. | ||||||
| 1 | n Pentyl | 3ba | 20 | 71 | >95:5 |
97 |
| 2 | Cyclohexyl | 3ca | 20 | 75 | >95:5 |
95 |
| 3 | o-BrPh | 3da | 20 | 79 | >95:5 |
98 |
| 4 | o-F3CPh | 3ea | 20 | 73 | >95:5 |
95 |
| 5 | 2-Pyridyl | 3fa | 28 | 68 | >95:5 |
98 |
| 6 | o-MePh | 3ga | 48 | 70 | >95:5 |
98 |
| 7 | o-MeOPh | 3ha | 72 | 64 | >95:5 |
98 |
| 8 | 2-Furyl | 3ia | 240 | 32 | >95:5 |
95 |

Mechanistic hypothesis
Our current working hypothesis is that the high enantioselectivity originates from a H-bond stabilised transition state (Scheme 2). This six-membered transition state consists of two H-bonds between the thiourea and nitro group as well as an amide H-bond with the iminium species. | ||
| Scheme 2 Origin of enantioselectivity. | ||
If we assume from the detailed work of Jacobsen on a similar catalyst that the most stable conformation of catalyst 5h is when the highlighted C–H bond is in the same plane as the large C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond,19 the amide can freely H-bond to the iminium species in a pseudo equatorial position (TS-1) giving rise to the observed enantiomer. Conversely, to obtain the opposite enantiomer the iso-propyl group would be required to be in the same plane as the CS bond (TS-2). Such a conformation would have a large steric penalty and would as such be unfavorable.
S bond,19 the amide can freely H-bond to the iminium species in a pseudo equatorial position (TS-1) giving rise to the observed enantiomer. Conversely, to obtain the opposite enantiomer the iso-propyl group would be required to be in the same plane as the CS bond (TS-2). Such a conformation would have a large steric penalty and would as such be unfavorable.
Preliminary investigations into forming three contiguous stereocentres
Pleased with the results using simple nitrostyrene derivatives we wished to examine the potential of this reaction to form nitro-Mannich products containing three contiguous stereocentres. Using nitroalkene 6 and our thiourea catalyst 5h we were pleasingly able to obtain 7a in high enantiopurity (84% ee) and excellent diastereoselectivity (95:0:5:0 dr) after 17 h at room temperature, albeit in a low yield of 37% (Table 5, entry 3). Interestingly, the reaction initially favoured formation of the syn, anti-diastereomer 7b before converting to 7a. We are currently unsure as to the mechanism of this interconversion but believe it may be formed via a retro addition/nitro-Mannich sequence.20 Further investigations and optimisations of this intriguing reaction are currently underway.

Conclusions
We have developed the first asymmetric tandem reductive nitro-Mannich reaction using a simple thiourea organocatalyst and a Hantzsch ester as hydride source. The reaction uses the simplest thiourea organocatalyst applied to a nitro-Mannich reaction yet and in general does not suffer from the long reaction times or large excess of reagents required of previous reports. The reductive nitro-Mannich strategy provides an opportunity for more diverse substrates and the reaction is tolerant of a variety of different nitroalkenes and imines. To the best of our knowledge this is also the first organocatalysed nitro-Mannich reaction using PMP-imines, which significantly simplifies the experimental conditions of the reaction and should enable wider use in synthesis. We are currently beginning detailed experimental and computational investigations into the origin of the exceptionally high levels of enantioselectivity considering the simplicity of the catalyst.Acknowledgements
Financial support was provided by the EPSRC (DTG) and UCL, we thank Dr L. Harris and Mr J. Hill for mass spectra, and Ms J. Maxwell for microanalytical data.Notes and references
- (a) V. Khama and S. Ranganathan, BMC Bioinformatics, 2009, 10, S10 CrossRef; (b) D. O'Hagan, Modern Alkaloids: Structure, Isolation, Synthesis and Biology, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (c) J. P. Michael, Nat. Prod. Rep., 2008, 25, 139 RSC and references there in.
- (a) R. Ballini and M. Petrini, Tetrahedron, 2004, 60, 1017 CrossRef CAS; (b) C. Palomo, M. Oiarbibe, R. Halder, A. Laso and R. López, Angew. Chem., Int. Ed., 2006, 45, 117 CrossRef CAS.
- (a) L. Bernadi, B. F. Bonini, I. E. Capito, G. Dessole, M. Comes-Franchimi, M. Fochi and A. Ricci, J. Org. Chem., 2004, 69, 8168 CrossRef; (b) H. Adams, J. C. Anderson, S. Peace and A. M. K. Pennel, J. Org. Chem., 1998, 63, 9932 CrossRef CAS; (c) D. H. Lloyd and D. E. Nichols, J. Org. Chem., 1986, 51, 4294 CrossRef CAS; (d) A. G. M. Barrett and C. D. Spilling, Tetrahedron Lett., 1988, 29, 5733 CrossRef CAS; (e) M. A. Sturgess and D. J. Yarberry, Tetrahedron Lett., 1993, 34, 4743 CrossRef CAS.
- M. G. Bursavich and D. H. Rich, J. Med. Chem., 2002, 45, 541 CrossRef CAS.
- (a) P. Jakubec, D. M. Cockfield and D. Dixon, J. Am. Chem. Soc., 2009, 131, 16632 CrossRef CAS; (b) P. Jakubec, A. F. Kyle, J. Calleja and D. J. Dixon, Tetrahedron Lett., 2011, 52, 6094 CrossRef CAS; (c) A. F. Kyle, P. Jakubec, D. M. Cockfield, E. Cleator, J. Skidmore and D. J. Dixon, Chem. Commun., 2011, 47, 10037 RSC; (d) B. Shen and J. N. Johnston, Org. Lett., 2008, 10, 4397 CrossRef CAS; (e) G. Kumaraswarmy and A. Pitchaiah, Helv. Chim. Acta, 2011, 94, 1543 CrossRef; (f) P. Jakubec, A. Hawkins, W. Felzmann and D. J. Dixon, J. Am. Chem. Soc., 2012, 134, 17482 CrossRef CAS.
- (a) N. Tsuritani, K.-I. Yamada, N. Yoshikawa and M. Shibasaki, Chem. Lett., 2002, 276 CrossRef CAS; (b) X. Xu, T. Furukawa, T. Okino, H. Miyabe and Y. Takemoto, Chem.–Eur. J., 2006, 12, 466 CrossRef; (c) P. S. Hynes, P. A. Stupple and D. J. Dixon, Org. Lett., 2008, 10, 1389 CrossRef CAS; (d) K. L. Jensen, P. H. Poulsen, B. S. Donslund, F. Morana and K. A. Jørgensen, Org. Lett., 2012, 14, 1516 CrossRef CAS; (e) S. Handa, V. Gnanadesikan, S. Matsunga and M. J. Shibasaki, J. Am. Chem. Soc., 2010, 132, 4925 CrossRef CAS; (f) J. Weng, Y.-B. Li, R.-B. Wang, F.-Q. Li, C. Liu, A. S. C. Chan and G. Lu, J. Org. Chem., 2010, 75, 3125 CrossRef CAS; (g) G. Kumaraswarmy and A. Pitchaiah, Tetrahedron, 2011, 67, 2536 CrossRef; (h) T. A. Davis and J. N. Johnston, Chem. Sci., 2011, 2, 1076 RSC; (i) T. A. Davis, M. W. Danneman and J. N. Johnston, Chem. Commun., 2012, 48, 5578 RSC; (j) H. Xie, Y. Zhang, S. Zhang, X. Chen and W. Wang, Angew. Chem., Int. Ed., 2011, 50, 11773 CrossRef CAS.
- (a) For a review specific to asymmetric methods see E. Marquéz-López, P. Merino, T. Tejero and R. P. Herrera, Eur. J. Org. Chem., 2009, 2401 CrossRef; (b) For a comprehensive review of the nitro-Mannich reaction see A. Noble and J. C. Anderson, Chem. Rev., 2013, 113, 2887 CrossRef CAS.
- Most examples require 5–10 equivalents of nitroalkane or several days to reach completion. Recently however, procedures have been disclosed which overcome this limitation: (a) T. A. Davis, J. C. Wilt and J. N. Johnston, J. Am. Chem. Soc., 2010, 132, 2880 CrossRef CAS; (b) W. Huang, C. Peng, L. Guo, R. Hu and B. Han, Synlett, 2011, 2981 CAS.
- R. A. Shenvi, D. P. O'Malley and P. S. Barran, Acc. Chem. Res., 2009, 42, 530 CrossRef CAS.
- (a) L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS; (b) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS; (c) I. Ibrahem and A. Córdova, Angew. Chem., Int. Ed., 2006, 45, 1952 CrossRef CAS; (d) S. Mukherjee and B. List, J. Am. Chem. Soc., 2007, 129, 11336 CrossRef CAS; (e) C. J. Chapman and C. G. Frost, Synthesis, 2007, 1 CAS; (f) A. M. Walji and D. W. C. MacMillan, Synlett, 2007, 1477 CAS; (g) Z.-Y. Han, H. Xiao, X.-H. Chenand and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 9182 CrossRef CAS; (h) H. Jiang, P. Elsner, K. L. Jensen, A. Falcicchio, V. Marcos and K. A. Jørgensen, Angew. Chem., Int. Ed., 2009, 48, 6844 CrossRef CAS; (i) S. Belot, K. A. Vogt, C. Besnard, N. Krause and A. Alexakis, Angew. Chem., Int. Ed., 2009, 48, 8923 CrossRef CAS.
- For a racemic reductive nitro-Mannich reaction see J. C. Anderson, A. J. Blake, P. J. Koovits and G. J. Stepney, J. Org. Chem., 2012, 77, 4711 CrossRef CAS.
- (a) For the most efficient anti-selective reactions see ref. 8a and C. Wang, X. Dong, Z. Zhang, Z. Xue and H. Teng, J. Am. Chem. Soc., 2008, 130, 8606 CrossRef CAS; (b) For the most efficient syn-selective reactions see S. Handa, V. Gnanadesikan, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 4925 CrossRef CAS.
- (a) T. Okino, S. Nakamura, T. Furukawa and Y. Takemoto, Org. Lett., 2004, 6, 625 CrossRef CAS; (b) T. P. Yoon and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 466 CrossRef CAS; (c) X. Xu, T. Furukawa, T. Okino, H. Miyabe and Y. Takemoto, Chem.–Eur. J., 2006, 12, 466 CrossRef.
- N. J. A. Martin, L. Ozores and B. List, J. Am. Chem. Soc., 2007, 129, 8976 CrossRef CAS.
- β-Nitroamines without electron withdrawing protecting groups on nitrogen are known to be unstable to standard purification techniques as they undergo retro-addition to re-form nitroalkane and imine.
- A possible explanation for the faster rate of reduction with tbutyl ester substituted Hantzsch ester has been forwarded: (a) S. G. Ouellet, A. Walji and D. W. C. Macmillan, Acc. Chem. Res., 2007, 40, 1327 CrossRef CAS; (b) J. B. Tuttle, PhD thesis, California Institute of Technology, 2008.
- Selected examples are shown in Scheme 2, for full catalyst screen see ESI.†.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 Search PubMed.
- For computational investigations with a similar catalyst see: S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2009, 131, 15358 Search PubMed.
- For a similar interconversion due to retro addition/re-addition nitro-Mannich reaction see: J. C. Anderson, G. J. Stepney, M. R. Mills, L. R. Horsfall, A. J. Blake and W. Lewis, J. Org. Chem., 2011, 76, 1961 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Method of determining absolute stereochemistry, detailed catalyst screen, full experimental details, 1H and 13C NMR spectra for all new compounds and HPLC data. See DOI: 10.1039/c3sc50613d |
| This journal is © The Royal Society of Chemistry 2013 |