 Open Access Article
Open Access ArticleSynthesis of spongistatin 2 employing a new route to the EF fragment†
Helmut
Kraus
a,
Antoine
Français
a,
Matthew
O'Brien
ab,
James
Frost
a,
Alejandro
Diéguez-Vázquez
a,
Alessandra
Polara
a,
Nikla
Baricordi
a,
Richard
Horan
a,
Day-Shin
Hsu
a,
Takashi
Tsunoda
a and
Steven V.
Ley
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: svl1000@cam.ac.uk; Fax: +44 (0)1223 336442; Tel: +44 (0)1223 336398
bLennard-Jones Laboratories, Keele University, Keele, Staffordshire ST5 5BG, UK. E-mail: m.obrien@keele.ac.uk; Tel: +44 (0)1782 734371
First published on 7th March 2013
Abstract
An improved route to the EF fragment of the spongistatins has been developed and employed in a synthesis of spongistatin 2. The C48–C51 diene side chain, which lacks the chlorine substituent present in spongistatin 1, presented some compatibility issues during target assembly. These were overcome by implementing a late stage Stille cross coupling to construct the diene portion of the natural product.
Introduction
According to recent WHO figures, cancer is the 4th largest cause of death worldwide, accounting for 13% of total mortality in 2008.1 With an increasingly aging population, the incidence of cancer is predicted to follow an upward trend.2 In the urgent battle against this disease, natural products have (directly or indirectly) delivered many important therapeutic advances and remain an invaluable resource in this respect.3 Since they were isolated independently by Pettit,4 Kitagawa5 and Fusetani6 in 1993, the spongistatins have attracted considerable scientific interest, owing principally to their exceptional antitumour activity against a variety of cell types. Indeed, all nine members of the spongistatin family have displayed remarkable growth inhibition characteristics against the US National Cancer Institute's panel of 60 human cancer cell lines.7 Of the family, spongistatin 1 is the most active, having GI50 values between 0.025–0.035 nM with particular efficacy against cell lines derived from human melanoma.7 Unfortunately, due to the very low abundance of these compounds, as well as the difficulties associated with isolation and purification, obtaining significant quantities of natural material for further research is neither practical nor economic. Consequently, significant research efforts have been made towards their total synthesis. The absolute stereochemistry of these complex 42 membered macrolides remained unknown until the first syntheses of spongistatin 2 by Evans8 in 1997 and spongistatin 1 by Kishi a year later.9 Since then, several additional total syntheses have been established by Crimmins,10 Heathcock,11 Paterson,12 Smith13 and ourselves.14For our total synthesis of spongistatin 1 we developed an efficient and scalable route to the ABCD fragment which profited from the pseudo-C2 symmetry of this fragment.14d It was clear, however, that despite successfully completing the total synthesis of spongistatin 1, access to biologically significant quantities of these natural products would require an improved synthetic route to the EF fragment; one which was more scalable, required fewer synthetic steps and provided a higher overall yield. We report herein our second generation synthesis of the EF fragment and its subsequent employment in a total synthesis of spongistatin 2.14e
Retrosynthesis
In line with previous syntheses of the spongistatins,8–14 our initial retrosynthetic analysis begins with disconnection of the C41(O)–C1 ester and the C28–C29 olefin to afford the ABCD aldehyde fragment, along with the corresponding EF phosphonium salt (3) (Fig. 1). As we already had a satisfactory route to the ABCD fragment (with material in hand),14d as well as a proven endgame strategy, we sought to use a similar protecting group regime for this new synthesis. However, whilst our earlier synthesis of spongistatin 1 used an EF fragment containing a fully assembled C48–C51 chlorodiene appendage, the nor-chlorodiene required for spongistatin 2 alerted us to potential compatibility concerns during the subsequent removal of the PMB protecting groups.11d Conjugated dienes are prone to undergo side reactions when exposed to DDQ,15 the standard reagent used for the oxidative cleavage of PMB groups, and it seems the chlorine substituent in spongistatin 1 mitigates this.12a,14d These concerns notwithstanding, we elected to continue with PMB protecting groups for the EF fragment. Experiments (including model studies) confirmed that the diene would likely not survive the conditions used to remove the PMB groups. For this reason we chose to append a vinyl iodide substituent which we hoped to transform at a late stage (after PMB deprotection) into the diene using a Stille coupling with tributyl(vinyl)tin.16 Further disconnection of EF fragment 3via two aldol reactions leads to trans vinyl iodide 4, F ring-containing fragment 5 and aldehyde 6 as potential coupling partners. Fragment 5 would then derive from the intramolecular oxy-Michael cyclisation of substrate 7. Development of a sequence capable of furnishing multigram quantities of fragment 5 was therefore key to a scalable route to the EF fragment and thereafter the completion of spongistatin 2 (2) (Fig. 1). | ||
| Fig. 1 Retrosynthetic analysis of spongistatin 2 and the EF fragment. | ||
Results and discussion
Our synthesis began from the commercially available (S)-Roche ester 8. This was protected as its p-methoxybenzyl ether, reduced to the corresponding aldehyde and reacted with Ph3PCHCO2Et to furnish enoate 9 in excellent yield as a single (E)-isomer. Dihydroxylation using the Sharpless17 AD-mix-β provided 10 with excellent diastereoselectivity (dr = 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6). Fortunately, this diol was highly crystalline and recrystallisation furnished a diastereomerically pure product in 92% yield. Protection as the acetonide followed by addition of MeLi·LiBr to the ester and Wittig reaction of the resulting ketone afforded 11. This 7 step sequence was carried out on large scale (100 mmol batches), allowing for the generation of 11 in substantial amounts. Removal of the PMB group, oxidation and Wittig–Horner reaction with known phosphonate 1318 yielded 14, which underwent selective dihydroxylation (dr = 86:14, single isomer after chromatography) to afford diol 15 (Scheme 1). We next focused on the construction of the F ring by means of an oxy-Michael cyclisation reaction.
:6). Fortunately, this diol was highly crystalline and recrystallisation furnished a diastereomerically pure product in 92% yield. Protection as the acetonide followed by addition of MeLi·LiBr to the ester and Wittig reaction of the resulting ketone afforded 11. This 7 step sequence was carried out on large scale (100 mmol batches), allowing for the generation of 11 in substantial amounts. Removal of the PMB group, oxidation and Wittig–Horner reaction with known phosphonate 1318 yielded 14, which underwent selective dihydroxylation (dr = 86:14, single isomer after chromatography) to afford diol 15 (Scheme 1). We next focused on the construction of the F ring by means of an oxy-Michael cyclisation reaction.
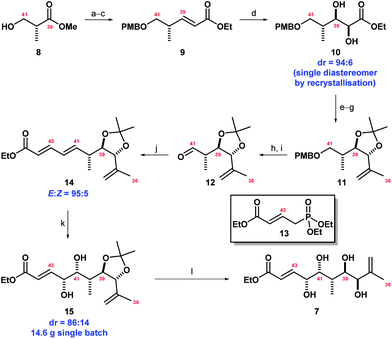 | ||
| Scheme 1 Synthesis of fragment 7. Reagents and conditions: (a) PMB-trichloroacetimidate, PPTS, CH2Cl2, r.t., quant.; (b) DIBAL-H, CH2Cl2, −78 °C; (c) Ph3PCHCO2Et, CH2Cl2, r.t., 94% over 2 steps; (d) AD-mix-β, MeSO2NH2, t-BuOH–H2O (1:1), 4 °C, 92%; (e) 2,2-dimethoxypropane, CSA, r.t., 96%; (f) MeLi·LiBr, Et2O, −78 °C, 95%; (g) CH3PPh3Br, n-BuLi, THF, 0 °C to r.t., 96%; (h) DDQ, pH 7 buffer, CH2Cl2, r.t.; (i) Dess–Martin periodinane, CH2Cl2, 0 °C to r.t.; (j) 13, LiHMDS, THF, −78 °C then 12, −78 °C to r.t.; 80% over 3 steps; (k) AD-mix-β, MeSO2NH2, t-BuOH–H2O (1:1), 0 °C, 89%; and (l) MP-TsOH, EtOH–H2O (1:1), 82 °C, 98%. | ||
Encouraged by studies undertaken by Paterson and co-workers,12d our initial efforts centred on removal of the acetonide protecting group in 15 under acidic conditions, which we hoped would be accompanied by spontaneous cyclisation to form the F ring in a one-pot procedure. However, although the acetonide could cleanly be removed (using MP-TsOH), the resulting tetraol 7 was resistant to subsequent cyclisation, despite assaying a range of conditions for this transformation (inc. Amberlyst A-15, TFA, Tf2NH, TfOH, AcOH, TMSCl:EtOH (0.3 M)). This being the case, we focussed our attention on the cyclisation process itself. The use of a variety of different bases (TBAF, NaH, KHMDS, t-BuOK, DBU, Et3N, NaHCO3) under different conditions (solvent and temperature) either failed to promote any reaction or afforded the desired product only in low yield (≤50%).19 In many instances the desired thermodynamic pyran 16 was formed in varying ratio with products resulting from epimerisation at C42 or C43, the latter of which tended to form the 6,5-cis fused bicyclic lactone 17. Having achieved little success with bases, we turned instead to the use of Lewis acids in order to affect this crucial transformation. Although such methods to construct densely functionalised tetrahydropyran rings are not widely reported,20 it seemed reasonable that a Lewis acid might activate the enoate towards nucleophilic attack. A selection of the additives which were screened are shown in Table 1.
|
|
||||
|---|---|---|---|---|
| Entry | Lewis acid | Temp. [°C] | Methoda | Result |
| a Method A = conventional heating using a reflux condenser; Method B = microwave heating in a sealed vessel; Method C = sealed Schlenk tube. b X = F, Br, I. c The reaction was also conducted at r.t. at elevated pressure (50 bar) with no conversion observed. | ||||
| 1 | TiCl4 | 80 | A | Decomposition |
| 2 | PtCl4 | 80 | A | No reaction |
| 3 | AuCl3 | 80 | A | No reaction |
| 4 | CeCl3 | 80 | A | No reaction |
| 5 | ZnCl2 | 80 | A | No reaction |
| 6 | PtCl4 | 110 | B | Decomposition |
| 7 | AuCl3 | 140 | B | Decomposition |
| 8 | CeCl3 | 120 | B | Decomposition |
| 9 | ZnCl2 | 180 | B | 16 (65%) + 17 (15%) |
| 10b | ZnX2 | 140 | B | Decomposition |
| 11 | Zn(OTf)2 | 100 | B | 16 (75%) + 17 (15%) |
| 12 | Zn(OTf)2 | 100 | C | 16 (88%) + 17 (8%)c |
| 13 | TfOH | 100 | C | No reaction |

The presence of a variety of Lewis acids (20 mol%) at 80 °C in ethanol either led to no reaction or to decomposition (entries 1–5). The results for Pt(IV) and Au(III) were disappointing as we speculated that the propensity of these metals to activate ynones toward intramolecular reactions would extend to enones.21 In order to investigate the use of higher temperatures with the same solvent, a microwave reactor was used to conveniently allow the solvent to be superheated under pressure (ethanol b.p. = 78.4 °C at 1 atm) (entries 6–11).22 Extensive decomposition was observed in several cases (entries 6–8). However, ZnCl2, which had no effect at 80 °C, afforded at 180 °C the desired product 16 in 65% yield, in addition to lactone 17 (15%). Whilst the use of other zinc(II) halogenide additives (ZnF2, ZnBr2, ZnI2) were ineffective (entry 10), Zn(OTf)2 proved to be the optimal choice, with the desired product produced in 75% yield (entry 11). Fortunately, this reaction proved to be both higher yielding (88%) and more scalable when performed in a sealed Schlenk tube, using conventional heating (oil bath) at 100 °C. Negating the need for a microwave reactor allowed multigram batches of material to be processed (entry 12). The origins of the superiority of Zn(OTf)2 as a catalyst for this reaction are not clear, although it seems unlikely that any residual TfOH is acting as a catalyst (entry 13).
Having developed an effective procedure to ensure scalable and efficient access to the F-ring scaffold we required a method that would allow for the protection of all three hydroxyl groups present in 16. PMB protection of hydroxyl groups is typically achieved using one of two sets of conditions: (a) base in conjunction with PMB-Cl/Br and a catalytic amount of TBAI/NaI23 or (b) PMB-trichloroacetimidate (TCA) combined with a Brønsted or Lewis acid catalyst.24 The latter method was investigated initially since experience gained from the preceding oxy-Michael cyclisation had revealed the C43 stereocentre to be highly prone to epimerisation. However, despite trying a variety of different acid catalysts (TfOH, La(OTf)3, BF3·OEt2, Ph3CBF4) these efforts were met with decomposition or only low yields of the desired tris-PMB ether 18. Little improvement was observed using tried-and-tested basic conditions. Wishing to avoid the daunting prospect of reworking our protecting group strategy, we persevered. After investigating a plethora of reagents and workarounds, we were pleased to find that a respectable 72% yield of 18 could be obtained using PMB-lepidine in the presence of MeOTs.25 Use of this reagent offered several advantages compared to PMB-TCA: (a) it was stable at room temperature and could be easily prepared,25 (b) the reactive intermediate could be formed under non-acidic conditions and (c) the lepidine byproduct could be easily removed by silica gel chromatography.
Hydrolysis of the ester functionality and formation of the corresponding Weinreb amide26 proceeded smoothly. Thereafter dihydroxylation of the alkene and Pb(OAc)4-mediated cleavage of the resulting diols furnished ketone 5; allowing for interception of our first generation synthesis of spongistatin 1 (Scheme 2).14e
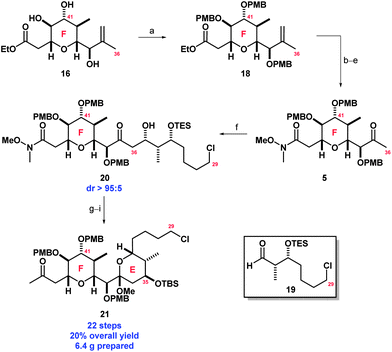 | ||
| Scheme 2 Synthesis of EF fragment 21. Reagents and conditions: (a) PMBO-lepidine, MeOTs, PhMe, 40 °C, 72%; (b) LiOH·H2O, THF, H2O, r.t.; (c) HCl·HN(OMe)Me, HOBt–EDCI, Et3N, THF, r.t, 86% over 2 steps; (d) OsO4, NMO, acetone–H2O (1:1), r.t., 98%; (e) Pb(OAc)4, PhMe, r.t., 98%; (f) cHex2BCl, Et3N, Et2O, 0 °C, 2 h, then 19, −78 °C, 2 h, 82%; (g) PPTS, CH(OMe)3, MeOH, r.t., 89%; (h) TBSOTf, 2,6-lutidine, THF, −78 °C, 99%; and (i) MeLi, CeCl3, −78 °C, quant. | ||
With ketone 5 in hand, our efforts shifted to the construction of the E ring. The boron mediated aldol coupling of ketone 5 and aldehyde 1927 was achieved in high yield (82%) and in excellent diastereoselectivity (dr > 95:5), giving the Felkin–Anh product (20).28 Presumably the 1,2-stereochemical induction predicted by the Felkin–Anh model overrides any 1,3-asymmetric induction favoured by the chiral aldehyde.29 Treatment of 20 with PPTS in the presence of trimethyl orthoformate and MeOH effected C33(OH) silyl deprotection and triggered spontaneous E ring cyclisation with concomitant formation of the C37 methyl acetal. The C35(OH) was then protected as its TBS ether, and the Weinreb amide functionality converted to the corresponding ketone (21) in quantitative yield by reaction with a methylcerium reagent prepared in situ from methyllithium and anhydrous ceric chloride.30 Completion of EF fragment 21via this new route resulted in a significantly shorter reaction sequence (22 linear steps) and a much higher overall yield of 20% compared to our first generation synthesis (27 linear steps and 5.6% overall yield).14e This enabled the production of 6.4 g of highly functionalised compound 21, and compares favourably with other routes reported towards similar EF fragments.12,13
At this stage the route diverges from our original spongistatin 1 synthesis. Aldol coupling of 21 with an excess31 of 3-iodo-acrolein 2232 furnished compound 23 in high diastereoselectivity (dr > 95:5) and good yield (65%) (Scheme 3).33 This was then TBS-protected, and subjected to the Takai–Lombardo methylenation34 conditions followed by Finkelstein reaction35 in a highly efficient 3 step sequence in 74% overall yield. Following considerable experimentation, a Wittig olefination reaction between the corresponding phosphonium salt 3 (1.0 equiv.) and ABCD aldehyde 24 (1.3 equiv.)14d in the presence of LiHMDS (3.0 equiv.) at −78 °C resulted in a 56% yield of the (Z)-alkene. The three PMB protecting groups in this compound were then oxidatively cleaved in 71% yield using DDQ.
 | ||
| Scheme 3 Completion of spongistatin 2. Reagents and conditions: (a) Cy2BCl, Et3N, Et2O, −55 °C, then 22, −70 °C, Et2O, 65%; (b) TBSCl, imidazole, DMF, r.t.; (c) Zn, PbI2, TMSCl, CH2I2, 0 °C to r.t., then TiCl4, CH2Cl2, r.t., followed by ketone, THF, r.t.; (d) NaI, NaHCO3, Na2SO3, 1-iodopropane, acetone, 67 °C, 74% over 3 steps; (e) PPh3, DIPEA, MeCN, 90 °C; (f) 3, THF, r.t. then LiHMDS, −78 °C, followed by aldehyde 24, −78 °C, 56% over 2 steps; (g) DDQ, pH 7 phosphate buffer, CH2Cl2, 0 °C, 71%; (h) Pd2dba3 (10 mol%), AsPh3(40 mol%), tributyl(vinyl)tin (5.0 equiv.), morpholine (10.0 equiv.), DMF–THF (1:1), r.t., 84%; (i) 2,4,6-trichlorobenzoyl chloride, DIPEA, DMAP, PhMe, 90 °C; and (j) 47–51% aq. HF, MeCN, −20 °C, 42% over 2 steps. | ||
At this stage the C48–C51 diene side chain could be completed. Pleasingly, the necessary Stille cross coupling reaction was not perturbed by the C1 allyl ester protecting group. A one-pot Stille-coupling/deprotection sequence36 was effected using Pd2dba3 (10 mol%), AsPh3(40 mol%), tributyl(vinyl)tin (5.0 equiv.) and morpholine (10.0 equiv.), affording 26 in an excellent yield of 84%. By installing the diene unit at this late stage, we effectively avoided the side reactions (oxidation, cycloadduct formation11d), that we and others had previously observed using DDQ on a similar diene substrate.11d Yamaguchi macrolactonisation37 of seco-acid 26 using the conditions developed by Heathcock11 led to a mixture of the desired product as well as some byproducts. From the 1H NMR of the crude product mixture, it appeared that partial hydrolysis of the methyl acetal at C37 had occurred, somewhat hampering full 1H NMR analysis. However, treatment of the crude mixture with 47–51% aq. HF at −20 °C resulted in global silyl deprotection and methyl acetal hydrolysis to deliver spongistatin 2 (2) in 42% over the two steps. The synthetic material was found to be identical to the natural product in all respects (1H NMR, 13C NMR, IR, mass spectrometry, optical rotation).4b,5c
Conclusions
In summary we have developed a highly scalable and efficient preparation of the EF fragment of the spongistatins. This was made possible by the design of a route that gave access to multigram quantities of tetraol 7. Satisfactory conditions for the F ring cyclisation initially proved elusive but this key transformation was eventually achieved in a yield of 88% using Zn(OTf)2. Further synthetic manipulation brought about interception with our first generation synthesis, leading to compound 21 in 22 steps (longest linear sequence) and 20% overall yield. With 5 fewer steps and a greater than threefold increase in yield, this clearly represents a significant gain in efficiency. A boron mediated aldol reaction using 3-iodo-acrolein (22) allowed us to install a truncated vinyl iodide side chain. By extending this to the diene at a late stage via a Stille cross coupling with tributyl(vinyl)tin, we managed to avoid serious incompatibility issues related to PMB deprotection and were able to complete a total synthesis of spongistatin 2 (2) with an overall yield of 1.3% (from Roche ester) over 32 steps (longest linear sequence). To date, 6.4 g of 21 have been prepared. With scalable routes to both the EF and ABCD fragments at our disposal, we are now in a position to synthesise spongistatin 2 (and other members of the family) in quantities useful for further biological investigation.Acknowledgements
We gratefully acknowledge the Cambridge European Trust for a partially funded studentship (H.K.), the EPSRC (A.F., M.O.B., R.H. grant number: EP/F0693685/1), Novartis (J.F., A.P.), the Ministerio de Educación y Ciencia (Spain) for a postdoctoral fellowship (A.D.V), Università di Ferrara (N.B.) the Taiwan Merit Scholarship Program for a postdoctoral fellowship (D.S.H.), Astellas Pharma Inc. (T.T.) and the B.P. 1702 endowment (S.V.L.) for generous funding.Notes and references
- http://www.who.int, accessed 01 February 2013.
- M. Mistry, D. M. Parkin, A. S. Ahmad and P. Sasieni, Br. J. Cancer, 2011, 105, 1795 CrossRef CAS.
- (a) M. Gordaliza, Clin. Transl. Oncol., 2007, 9, 767 CrossRef CAS; (b) G. M. Cragg, P. G. Grothaus and D. J. Newman, Chem. Rev., 2009, 109, 3012 CrossRef CAS; (c) A. L. Demain and P. Vaishnav, Microb. Biotechnol., 2011, 4, 687 CrossRef.
- (a) G. R. Pettit, Z. A. Cichacz, F. Gao, C. L. Herald, M. R. Boyd, J. M. Schmidt and J. N. A. Hooper, J. Org. Chem., 1993, 58, 1302 CrossRef CAS; (b) G. R. Pettit, Z. A. Cichacz, F. Gao, C. L. Herald and M. R. Boyd, J. Chem. Soc., Chem. Commun., 1993, 1166 RSC; (c) G. R. Pettit, C. L. Herald, Z. A. Cichacz, F. Gao, J. M. Schmidt, M. R. Boyd, N. D. Christie and F. E. Boettner, J. Chem. Soc., Chem. Commun., 1993, 1805 RSC; (d) G. R. Pettit, C. L. Herald, Z. A. Cichacz, F. Gao, M. R. Boyd, N. D. Christie and J. M. Schmidt, Nat. Prod. Lett., 1993, 3, 239 CrossRef CAS; (e) G. R. Pettit, Z. A. Cichacz, C. L. Herald, F. Gao, M. R. Boyd, J. M. Schmidt, E. Hamel and R. L. Bai, J. Chem. Soc., Chem. Commun., 1994, 1605 RSC.
- (a) M. Kobayashi, S. Aoki, H. Sakai, K. Kawazoe, N. Kihara, T. Sasaki and I. Kitagawa, Tetrahedron Lett., 1993, 34, 2795 CrossRef CAS; (b) M. Kobayashi, S. Aoki and I. Kitagawa, Tetrahedron Lett., 1994, 35, 1243 CrossRef CAS; (c) M. Kobayashi, S. Aoki, H. Sakai, N. Kihara, T. Sasaki and I. Kitagawa, Chem. Pharm. Bull., 1993, 41, 989 CrossRef CAS.
- N. S. Fusetani, K. Shinoda and S. Matsunaga, J. Am. Chem. Soc., 1993, 115, 3977 CrossRef CAS.
- (a) R. L. Bai, Z. A. Cichacz, C. L. Herald, G. R. Pettit and E. Hamel, Mol. Pharmacol., 1993, 44, 757 CAS; (b) R. L. Bai, G. F. Taylor, Z. A. Cichacz, C. L. Herald, J. A. Kepler, G. R. Pettit and E. Hamel, Biochemistry, 1995, 34, 9714 CrossRef CAS; (c) R. K. Pettit, S. C. McAllister, G. R. Pettit, C. L. Herald, J. M. Johnson and Z. A. Cichacz, Int. J. Antimicrob. Agents, 1998, 9, 147 CrossRef CAS.
- (a) D. A. Evans, P. J. Coleman and L. C. Dias, Angew. Chem., Int. Ed., 1997, 36, 2738 CrossRef CAS; (b) D. A. Evans, B. W. Trotter, B. Côté and P. J. Coleman, Angew. Chem., Int. Ed., 1997, 36, 2741 CrossRef CAS; (c) D. A. Evans, B. W. Trotter, B. Côté, P. J. Coleman, L. C. Dias and A. N. Tyler, Angew. Chem., Int. Ed, 1997, 36, 2744 CrossRef CAS; (d) D. A. Evans, B. W. Trotter, P. J. Coleman, B. Côté, L. C. Dias, H. A. Rajapakse and A. N. Tyler, Tetrahedron, 1999, 55, 8671 CrossRef CAS.
- (a) J. S. Guo, K. J. Duffy, K. L. Stevens, P. I. Dalko, R. M. Roth, M. M. Hayward and Y. Kishi, Angew. Chem., Int. Ed., 1998, 37, 187 CrossRef CAS; (b) M. M. Hayward, R. M. Roth, K. J. Duffy, P. I. Dalko, K. L. Stevens, J. S. Guo and Y. Kishi, Angew. Chem., Int. Ed., 1998, 37, 192 CrossRef CAS.
- (a) M. T. Crimmins and D. G. Washburn, Tetrahedron Lett., 1998, 39, 7487 CrossRef CAS; (b) M. T. Crimmins and J. D. Katz, Org. Lett., 2000, 2, 957 CrossRef CAS; (c) M. T. Crimmins, J. D. Katz, D. G. Washburn, S. P. Allwein and L. F. McAtee, J. Am. Chem. Soc., 2002, 124, 5661 CrossRef CAS.
- (a) M. M. Claffey, C. J. Hayes and C. H. Heathcock, J. Org. Chem., 1999, 64, 8267 CrossRef CAS; (b) G. A. Wallace, R. W. Scott and C. H. Heathcock, J. Org. Chem., 2000, 65, 4145 CrossRef CAS; (c) J. L. Hubbs and C. H. Heathcock, J. Am. Chem. Soc., 2003, 125, 12836 CrossRef CAS; (d) C. H. Heathcock, M. McLaughlin, J. Medina, J. L. Hubbs, G. A. Wallace, R. Scott, M. M. Claffey, C. J. Hayes and G. R. Ott, J. Am. Chem. Soc., 2003, 125, 12844 CrossRef CAS.
- (a) I. Paterson, D. Y. K. Chen, M. J. Coster, J. L. Acena, J. Bach, K. R. Gibson, L. E. Keown, R. M. Oballa, T. Trieselmann, D. J. Wallace, A. P. Hodgson and R. D. Norcross, Angew. Chem., Int. Ed., 2001, 40, 4055 CrossRef CAS; (b) I. Paterson, M. J. Coster, D. Y.-K. Chen, R. M. Oballa, D. J. Wallace and R. D. Norcross, Org. Biomol. Chem., 2005, 3, 2399 RSC; (c) I. Paterson, M. J. Coster, D. Y.-K. Chen, K. R. Gibson and D. J. Wallace, Org. Biomol. Chem., 2005, 3, 2410 RSC; (d) I. Paterson, M. J. Coster, D. Y.-K. Chen, J. L. Aceña, J. Bach, L. E. Keown and T. Trieselmann, Org. Biomol. Chem., 2005, 3, 2420 RSC; (e) I. Paterson, D. Y.-K. Chen, M. J. Coster, J. L. Aceña, J. Bach and D. J. Wallace, Org. Biomol. Chem., 2005, 3, 2431 RSC.
- (a) A. B. Smith, III, V. A. Doughty, Q. Lin, L. Zhuang, M. D. McBriar, A. M. Boldi, W. H. Moser, N. Murase, K. Nakayama and M. Sobukawa, Angew. Chem., Int. Ed., 2001, 40, 191 CrossRef; (b) A. B. Smith, III, Q. Y. Lin, V. A. Doughty, L. H. Zhuang, M. D. McBriar, J. K. Kerns, C. S. Brook, N. Murase and K. Nakayama, Angew. Chem., Int. Ed., 2001, 40, 196 CrossRef; (c) A. B. Smith, III, A. Doughty, C. Sfouggatakis, C. S. Bennett, J. Koyanagi and M. Takeuchi, Org. Lett., 2002, 4, 783 CrossRef; (d) A. B. Smith, III, W. Zhu, S. Shirakami, C. Sfouggatakis, V. A. Doughty, C. S. Bennett and Y. Sakamoto, Org. Lett., 2003, 5, 761 CrossRef; (e) A. B. Smith, III, Q. Y. Lin, V. A. Doughty, L. H. Zhuang, M. D. McBriar, J. K. Kerns, A. M. Boldi, N. Murase, W. H. Moser, C. S. Brook, C. S. Bennett, K. Nakayama, M. Sobukawa and R. E. L. Trout, Tetrahedron, 2009, 65, 6470 CrossRef; (f) A. B. Smith, III, C. Sfouggatakis, C. A. Risatti, J. B. Sperry, W. Y. Zhu, V. A. Doughty, T. Tomioka, D. B. Gotchev, C. S. Bennett, S. Sakamoto, O. Atasoylu, S. Shirakami, D. Bauer, M. Takeuchi, J. Koyanagi and Y. Sakamoto, Tetrahedron, 2009, 65, 6489 CrossRef.
- (a) E. Fernandez-Megia, N. Gourlaouen, S. V. Ley and G. J. Rowlands, Synlett, 1998, 991 CrossRef CAS; (b) M. J. Gaunt, D. F. Hook, H. R. Tanner and S. V. Ley, Org. Lett., 2003, 5, 4815 CrossRef CAS; (c) M. J. Gaunt, A. S. Jessiman, P. Orsini, H. R. Tanner, D. F. Hook and S. V. Ley, Org. Lett., 2003, 5, 4819 CrossRef CAS; (d) M. Ball, M. J. Gaunt, D. F. Hook, A. S. Jessiman, S. Kawahara, P. Orsini, A. Scolaro, A. C. Talbot, H. R. Tanner, S. Yamanoi and S. V. Ley, Angew. Chem., Int. Ed., 2005, 44, 5433 CrossRef CAS; (e) For our 1st generation synthesis of the EF fragment see: S. Kawahara, M. J. Gaunt, A. Scolaro, S. Yamanoi and S. V. Ley, Synlett, 2005, 2031 CAS; (f) M. O'Brien, A. Diéguez-Vázquez, D.-S. Hsu, H. Kraus, Y. Sumino and S. V. Ley, Org. Biomol. Chem., 2008, 6, 1159 RSC.
- (a) I. Paterson, C. J. Cowden, V. S. Rahn and M. D. Woodrow, Synlett, 1998, 915 CrossRef CAS; (b) S. M. Bauer and R. W. Armstrong, J. Am. Chem. Soc., 1999, 121, 6355 CrossRef CAS; (c) T. Onoda, R. Shirai and S. Iwasaki, Tetrahedron Lett., 1997, 38, 1443 CrossRef CAS.
- (a) D. Azarian, S. S. Dua, C. Eaborn and D. R. M. Walton, J. Organomet. Chem., 1976, 117, C55 CrossRef CAS; (b) M. Kosugi, K. Sasazawa, Y. Shimizu and T. Migita, Chem. Lett., 1977, 301 CrossRef CAS; (c) D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1978, 100, 3636 CrossRef CAS; (d) P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704 CAS and all references cited therein.
- (a) G. Li, H.-T. Chang and K. B. Sharpless, Angew. Chem., Int. Ed., 1996, 35, 451 CrossRef CAS; (b) O. Reiser, Angew. Chem., Int. Ed., 1996, 35, 1308 CrossRef CAS.
- B. Bennacer, D. Trubuil, C. Rivalle and D. S. Grierson, Eur. J. Org. Chem., 2003, 4561 CrossRef CAS.
- G. C. Micalizio, A. N. Pinchuk and W. R. Roush, J. Org. Chem., 2000, 65, 8730 CrossRef CAS.
- L. Dheilly, C. Lievre, C. Frechou and G. Demailly, Tetrahedron Lett., 1993, 34, 5895 CrossRef CAS.
- (a) A. Diéguez-Vázquez, C. C. Tzschucke, J. Crecente-Campo, S. McGrath and S. V. Ley, Eur. J. Org. Chem., 2009, 11, 1698 CrossRef; (b) A. Diéguez-Vázquez, C. C. Tzschucke, W.-L. Yam and S. V. Ley, Angew. Chem., Int. Ed., 2008, 47, 209 CrossRef; (c) J. R. Frost, C. M. Pearson, T. N. Snaddon, R. A. Booth and S. V. Ley, Angew. Chem., Int. Ed., 2012, 51, 9366 CrossRef CAS.
- For recent discussion/refutation of non-thermal ‘microwave effects’ see: (a) M. Hosseini, N. Stiasni, V. Barbieri and C. O. Kappe, J. Org. Chem., 2007, 72, 1417 CrossRef CAS; (b) J. Raimer, E. Renard and D. Grande, Macromol. Chem. Phys., 2012, 213, 784 CrossRef; (c) B. T. Ergan and M. Bayramoglu, Ind. Eng. Chem. Res., 2011, 50, 6629 CrossRef CAS.
- D. R. Mootoo and B. Fraser-Reid, Tetrahedron, 1990, 46, 185 CrossRef CAS.
- N. Nakajima, K. Horita, R. Abe and O. Yonemitsu, Tetrahedron Lett., 1988, 29, 4139 CrossRef CAS.
- (a) K. W. C. Poon, S. E. House and G. B. Dudley, Synlett, 2005, 3142 CAS; (b) K. W. C. Poon and G. B. Dudley, J. Org. Chem., 2006, 71, 3923 CrossRef CAS; (c) E. O. Nwoye and G. B. Dudley, Chem. Commun., 2007, 1436 RSC; (d) C. A. Stewart, X. W. Peng and L. A. Paquette, Synthesis, 2008, 433 CAS.
- (a) S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815 CrossRef CAS; (b) J. I. Levin, E. Turos and S. M. Weinreb, Synth. Commun., 1982, 12, 989 CrossRef CAS.
- I. Paterson, J. M. Goodman and M. Isaka, Tetrahedron Lett., 1989, 30, 7121 CrossRef CAS.
- W. R. Roush, T. D. Bannister, M. D. Wendt, M. S. Van Nieuwenhze, D. J. Gustin, G. J. Dilley, G. C. Lane, K. A. Scheidt and W. J. Smith III, J. Org. Chem., 2002, 67, 4284 CrossRef CAS.
- D. A. Evans, M. J. Dart, J. L. Duffy and M. G. Yang, J. Am. Chem. Soc., 1996, 118, 4322 CrossRef CAS.
- T. Imamoto, Pure Appl. Chem., 1990, 62, 747 CrossRef CAS.
- A large excess of 3-iodo-acrolein 22 was required due to its poor solubility in Et2O.
- C. Meyer, I. Marek and J.-F. Normant, Synlett, 1993, 386 CrossRef CAS.
- The desired (S)-configuration of the C47-stereocentre was confirmed by Mosher's ester analysis: (a) J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1973, 95, 512 CrossRef CAS; (b) T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451 CrossRef CAS.
- (a) F. N. Tebbe, G. W. Parshall and G. S. Reddy, J. Am. Chem. Soc., 1978, 100, 3611 CrossRef CAS; (b) L. Lombardo, Tetrahedron Lett., 1982, 23, 4293 CrossRef CAS; (c) J. J. Eisch and A. Piotrowski, Tetrahedron Lett., 1983, 24, 2043 CrossRef CAS; (d) N. A. Petasis and E. I. Bzowej, J. Am. Chem. Soc., 1990, 112, 6392 CrossRef CAS; (e) K. Takai, T. Kakiuchi, Y. Kataoka and K. Utimoto, J. Org. Chem., 1994, 59, 2668 CrossRef CAS.
- H. Finkelstein, Ber. Dtsch. Chem. Ges., 1910, 43, 1528 CrossRef CAS.
- S. Phoenix, M. S. Reddy and P. Deslongchamps, J. Am. Chem. Soc., 2008, 130, 13989 CrossRef CAS.
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sc50304f |
| This journal is © The Royal Society of Chemistry 2013 |