 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzyme inhibition by metal complexes: concepts, strategies and applications
Kelly J.
Kilpin
and
Paul J.
Dyson
Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: paul.dyson@epfl.ch; Fax: +41 (0)21 693 98 85; Tel: +41-21-6939854
First published on 18th February 2013
Abstract
Metal complexes are increasingly being used to inhibit enzymes. The reasons for this increased interest arise from the special features that metal complexes offer, e.g. the facile construction of 3D architectures that tightly fill enzyme active sites increasing selectivity and the possibility of facile coordination to protein residues that enhances enzyme inhibition. In this review we classify the main modes of enzyme inhibition by metal-based complexes and correlate the enzyme inhibition activity to macroscopic properties such as anticancer activity.
Rosenberg's discovery of the anticancer properties of cisplatin, [cis-Pt(NH3)2Cl2],1 and its subsequent successful transition into the clinic, changed the way in which inorganic compounds were perceived in biological and medicinal chemistry. In the ensuing years thousands of structurally different metal-based compounds have been synthesised and evaluated for anti-cancer activity.2,3 However, and somewhat disappointingly, from this vast selection of compounds, only three (cisplatin, 1, and its descendants, carboplatin, 2, and oxaliplatin, 3, Fig. 1), have progressed into worldwide clinical use,4 although some new metal-based drugs are on the horizon.5,6 Amongst the main reasons for this poor success rate, with respect to both entering and successfully completing clinical trials, are the low water solubility of many compounds coupled to their high general toxicity. That is, in addition to acting on cancer cells, healthy cells are also damaged, resulting in the undesirable side effects often associated with chemotherapeutic treatments, and a consequence of designing drugs that damage DNA. However, as our understanding of the pathways involved in the onset and progression of cancer increases, and the fields of genomics and proteomics rapidly develop, the possibility to rationally design compounds to specifically target certain characteristics unique to the disease also increases.7–9
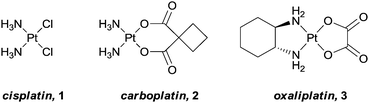 | ||
| Fig. 1 Platinum-based anticancer agents in worldwide clinical use. | ||
The ultimate cellular target of cisplatin (and other Pt-based clinical agents) is DNA,10 however in recent years other cellular components have emerged as possible targets for metal-based drugs.11–14 In particular, enzymes which play important roles in metabolic pathways associated with cancer are proving popular targets for metal-based drug design. This is not only true for cancer chemotherapy, but has been extended to other diseases of high social impact,15,16 and a number of reviews are dedicated toward metal-based enzyme inhibition, or more generally, protein inhibition.8,17–21
We propose that metal-based enzyme inhibitors can be classified into three main classes depending on the role the metal centre plays in the inhibition process:
(1) Complexes that contain a metal centre that does not directly coordinate to the enzyme. Instead, the ligands are the biologically active component and are responsible for protein binding/enzyme inhibition, although the metal may undergo redox events that enhance these effects.
(2) Complexes where the ligands merely mask a ‘naked’ metal ion, which usually, unselectively, interacts with many different protein targets. The ligands are usually not bioactive, but are present to stabilise or protect the reactive metal centre.
(3) Complexes where both the metal and the ligands are biologically active and are mutually responsible for the biological activity of the complex.
In this article we select examples of compounds that illustrate these three classifications, with an emphasis on complexes that have been rationally designed to selectively inhibit specific enzymes involved in cancer. For more comprehensive reviews readers are directed towards the aforementioned literature. In addition, metal complexes that have been designed to mimic enzyme activity, such a nucleases,22 proteases23 and superoxide dismutases (SODs)24 are not covered in this article.
Non-coordinating metal centres, active ligands
In pioneering studies, Jaouen successfully introduced redox active ferrocenyl fragments into the structure of known organic drugs.25,26 By replacing a phenyl ring on hydroxytamoxifen, 4, (the active metabolite of the tamoxifen – a potent drug used for the treatment of hormone dependent breast cancer) with a ferrocene unit, a new series of drugs (hydroxyferrocifens, 5, Fig. 2), were obtained. These ferrocenyl-derivatives demonstrate significantly different activity profiles to the original drug: tamoxifen is only active against breast tumours which exhibit estrogen receptors (ER(+)) whereas ferrocifens are also active towards tumours which lack this receptor (ER(−)). Consequently, the introduction of the metal fragment not only increases the treatment spectrum, but also overcomes problems associated with acquired resistance to tamoxifen.27,28 The proposed mechanism involves docking of the organic moiety into the receptor which is accompanied by two one-electron oxidations to give a quinone methide structure which is capable of forming adducts with biological nucleophiles.29 Such a mechanism is highly dependent on the redox properties of the ferrocene unit, thus the metal plays a role in the activity of the drug without coordinating to the receptor. The concept of introducing a ferrocenyl group was rapidly extended to the anti-malarial drug ferroquine, 7, (currently in phase III clinical trials), an analogue of chloroquine, 6, which is active against both chloroquine-sensitive and resistant parasites (Fig. 2).30 Here it is not clear if the metal ion is active or inert, nevertheless the modification results in uptake of the compound into resistant bacteria and is not easily detoxified, thus overcoming resistance. Although neither of these examples are active via enzyme inhibition, they deserve to be mentioned, as they elegantly demonstrate that the introduction of a metal fragment can significantly alter the activity profiles of known drugs, and to varying degrees, provided the motivation for many of the inhibitors that will be discussed in this article.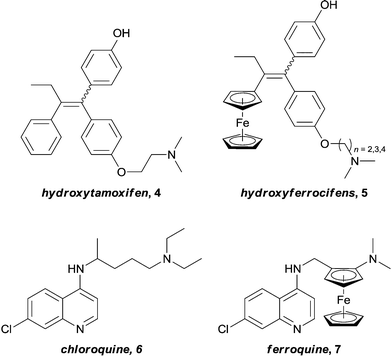
Two enzymes that are overexpressed in many types of cancer, Carbonic Anhydrase (CA, specifically the isoforms CA IX and CA XII)31,32 and Histone Deacetylase (HDAC),33 are zinc-containing metalloenzymes. In both enzymes the catalytic metal center is located at the end of a barrel shaped hydrophobic cavity.
Typical organic inhibitors of these enzymes comprise a zinc binding group (ZBG) such as a sulfonamide, a linker group which interacts with the hydrophobic cavity of the enzyme, and a tail group which is amenable to structural modification. Poulsen and co-workers have evaluated the benefit of introducing a metallocene, (either ferrocene or ruthenocene) onto the tail of CA inhibitors, separated from the ZBG (a chelating sulfonamide in this case) by either a 1,4-(8a, b) or a 1,5-1,2,3-triazole (9a, b) spacer (Fig. 3).34–36 X-ray crystallography of the complexes bound to the active site of human CA II (hCA II) confirmed the binding of the sulfonamide to the catalytic zinc, with the metallocene fragment located at the entrance to the cavity and apparently uninvolved in any interactions with the enzyme.35 Significantly, the compounds modified with metallocenes were generally superior inhibitors than the analogues containing a simple phenyl ring, and importantly, the metal-based compounds displayed higher selectivity towards cancer associated CA's over off-target CA's. The biopharmaceutical properties of the organic and metal-based drugs were shown to be similar, suggesting the enhanced activity and isoform discrimination of the metal-based drugs is due to the bulky metallocene and its ability to occupy important regions of the active site, inaccessible by more 2-dimensional organic groups.36 However, the authors (and indeed others, vide infra) point out that only slight perturbations to the metal group of the inhibitor can result in significant changes to the inhibition activity. For example, the ruthenocene complexes (8b, 9b), in which the two cyclopentadienyl (Cp) rings are slightly further apart than in ferrocene, are better inhibitors than the analogous ferrocene-containing species (8a, 9a) (Fig. 4).35
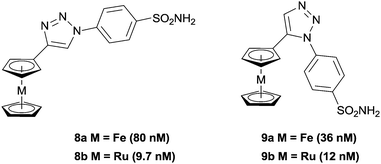 | ||
| Fig. 3 Ferrocene and ruthenocene 1,2,3-triazole inhibitors of Carbonic Anhydrase (CA), with inhibition constants (Kis) in parenthesis (hCA II). | ||
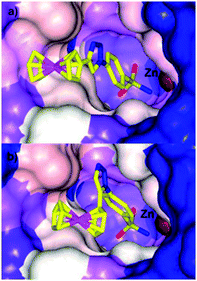 | ||
| Fig. 4 Comparison of the space filling in the active site of hCA II by (a) 8a (PDB 3P55) and (b) 9a (PDB 3P44) highlighting how a slight change in geometry can alter the fit in the active site.35 | ||
Alberto and co-workers have also investigated metal-based CA inhibitors, this time introducing rhenium and technetium tricarbonyl fragments onto the tail unit (Fig. 5).37 Like the ferrocene-containing inhibitors of Poulsen, a co-crystal structure of 11a with hCA II showed the sulfonamide group bound to the catalytically active zinc, and lacking interactions between the CpRe(CO)3 substituted tail group and the protein (Fig. 6). Interestingly, the compounds showed nanomolar affinities for particular CA subtypes, in particular compound 10 which showed enhanced selectivity for the isoforms hCA II, IX and XIV, a pattern of behaviour not observed for acetazolamide, a benchmark organic inhibitor. This approach was extended with the incorporation of technetium in place of rhenium (i.e. CpTc(CO)3, viz.11b) and as the inert 99mTc compounds exhibit biological behaviour identical to their rhenium counterparts this dyad has possible theranostic (i.e. therapeutic and diagnostic) applications.
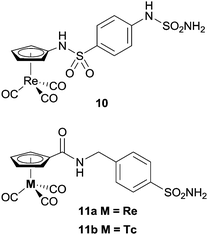 | ||
| Fig. 5 Examples of metal-carbonyl CA inhibitors. | ||
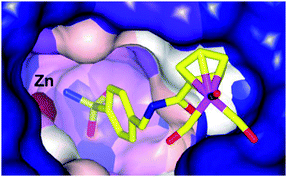 | ||
| Fig. 6 Binding of 11a to hCA II determined by X-ray crystallography (PDB 3RJ7), showing an intact Re coordination sphere and chelation of the active site zinc by the sulfonamide group.37 | ||
A co-crystal structure of the same protein, hCA II, with the ruthenium(II) piano stool complex 12c, bearing a sulfonamide zinc binding group has also been reported.38 The complex interacts with the protein in a manner reminiscent of the previously described inhibitors; the sulfonamide group binds to the catalytic zinc, the aryl spacer forms close contacts with the hydrophobic funnel shaped cavity and the ruthenium arene unit is positioned at the entrance of the cavity. There are no ruthenium–protein interactions and the ruthenium coordination sphere remains intact, despite the presence of a chloride leaving group. In accordance with previous observations, slight changes to the arene ring greatly affect the affinity of the complex for the enzyme, suggesting a subtle complementarity between the metal unit and the enzyme (Fig. 7).
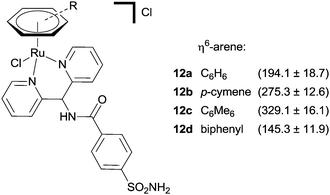 | ||
| Fig. 7 A series of piano-stool Ru(II) arene CA inhibitors with inhibition constants (nM, Kis) in parenthesis (hCA II). | ||
In a similar manner to that described for the CA inhibitors above, the clinically used HDAC inhibitor SAHA, 13, (Vorinostat) has been modified by replacement of the terminal phenyl ring with metal fragments (Fig. 8). Incorporation of a CpRe(CO)3 into the SAHA backbone resulted in compounds (e.g.14) that were cytotoxic against a range of cell lines (ca. 12 μM), albeit slightly less than SAHA (ca. 4 μM) itself, indicating that the introduction of such steric bulk promotes less favourable enzyme–inhibitor interactions (although the reduced cellular uptake of the metal-containing complex compared with SAHA cannot be discounted as a reason for the lower activity).39 Docking studies on the ferrocene modified species JAHA, 15, alluded to the molecule binding to the HDAC active site in a manner similar to SAHA.40 This proposition was subsequently corroborated when HDAC inhibition assays revealed SAHA and JAHA display similar nanomolar inhibition profiles against a selection of HDAC isozymes. As with the Re(CO)3 example, the cytotoxicities of JAHA, and other structurally modified JAHAs, were slightly less than that of SAHA itself.41
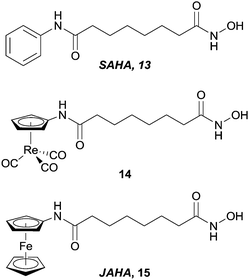 | ||
| Fig. 8 SAHA and metal-containing analogues evaluated as HDAC inhibitors. | ||
In contrast to modifying somewhat remote groups of organic inhibitors, Meggers et al. have taken the concept of using metal fragments to occupy defined regions of 3D-space in enzyme active sites one step further. As opposed to attaching an inert metal group to a known inhibitor, their concept was to use the inert metal as a scaffold in which to orientate biologically active ligands with well-defined topologies. Using this approach the metal complexes behave like organic molecules with the inert metal acting as a ‘hypervalent carbon’ atom with the metal centre placed at the heart of the inhibitor. With the increased coordination number and isomers available by virtue of the typically octahedral metal centre (compared with carbon), the complexes are able to occupy a defined region of 3D space, which results in a greater selectivity of the inhibitor relative to 2D organic structures.42
The lead compounds of this class were designed to mimic staurosporine, a complex natural product which acts as a potent ATP-competitive protein kinase inhibitor. The metal-based mimics retain the indolocarbazole core of staurosporine, which interacts with the cleft shaped kinase active site in a manner similar to ATP, but the carbohydrate moiety is replaced by an inert (typically) octahedral metal centre (Fig. 9). The resulting complexes are amenable to extensive structural modifications, e.g. by modifying the ancillary ligands around the metal.42
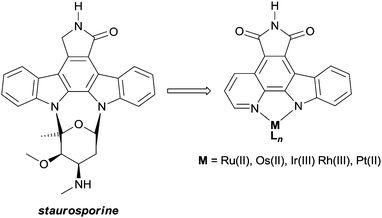 | ||
| Fig. 9 The natural product staurosporine and the metal-based mimics designed as protein kinase inhibitors. | ||
The initial complexes were shown to be inhibitors of Pim-1,43 and following a number of structural modifications (driven by structure based design, combinatorial chemistry, screening libraries etc.) inhibitors which display nanomolar, and in some cases picomolar kinase inhibitory properties, were obtained. Slight structural modifications resulted in substantial differences in selectivity towards different members of the kinase family – discriminating between different members of the family with organic molecules is problematic due to the highly conserved ATP binding sites.44–48
The biological impact of a slight modification on the ancillary ligand is illustrated in Fig. 10, which shows that minor modifications at an apparently remote site lead to large differences in inhibition values. This approach has been extended to other metal centres, including platinum,49 osmium,50 iridium51 and rhodium.52,53 In addition to a number of experiments used to demonstrate the inert character of the metal, a number of co-crystal structures have been determined, with the staurosporine mimics bound in the active site of the kinase enzyme (e.g.16a, Fig. 11).44–48,50,54–57 Regardless of the kinase, interactions between the ligand and the active site, which are reminiscent of ATP and staurosporine binding, are retained. In all cases the metal ion plays a purely structural role, and is not involved in any direct contacts with residues in the active site, and therefore it can be considered to act as a type of glue holding together different ligands. This approach also allows diversity to be easily introduced in the synthetic process, allowing libraries of related inhibitors to be generated and screened, facilitating the discovery of highly active and selective enzyme inhibitors.54,58,59
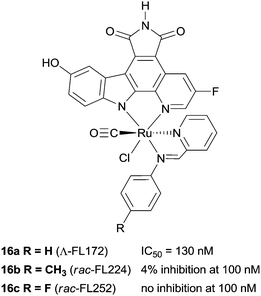 | ||
| Fig. 10 Examples of the protein kinase inhibitors with selected PAK-1 inhibition parameters (measured in the presence of 1 μM ATP).8 | ||
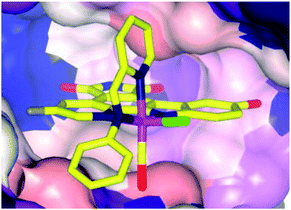 | ||
| Fig. 11 Co-crystal structure of 16a (Λ-FL172, PDB 3FXZ) and protein kinase PAK-1 with 16a bound at the ATP-binding site of the enzyme (C-terminal domain), showing the tight fit of 16a with the hinge region of the enzyme.44 | ||
Active metal centres
In the previous section, the examples describe cases where the metal plays a structural role, allowing the inhibitors to occupy a defined space in an enzymes' active site. At the opposite end of the spectrum are those complexes where the ligand is essentially biologically ‘innocent’, and the metal centre is entirely responsible for the activity of the complex. There are a plethora of examples in the literature of inorganic complexes where metal ions with simple ligands inhibit enzymes, especially relating to ‘soft’ metal ions, namely Pt(II), Ru(III), Au(I) and Au(III), that inhibit enzymes by binding with soft donor atoms on amino acid residues, typically the sulfur in cysteine residues, at both active and allosteric sites. In particular, a number of reviews describe the protein interactions of platinum and gold drugs, showing that such interactions are critical to their mode of action and that DNA is not the only relevant target.60,61 In some cases however, the increased liability of the metal ion allows a more complete exchange of the original ligands with binding residues on potential biomolecular targets, which in turn reduces selectivity and may account for the general toxicity of the compounds. Take for example relatively simple Au(I) thiolate complexes, such as auranofin, 17, which is used in the clinic to treat severe rheumatoid arthritis. Early studies on the anticancer activity of auranofin revealed activity levels similar to cisplatin in vitro, which subsequently led to a large number of Au(I) complexes being evaluated for anti-proliferative activity.62 Although the mechanism of action of these compounds is still not fully understood, inhibition of enzymes seems to play a major role.63 However, it appears that the compounds are non-selective, and investigations have revealed that such compounds not only inhibit a number of significant enzymes including thioredoxin reductase (TrxR),64 cysteine proteases,65 kinases66 and glutathione transferase (GST P1-1),67 but also interact with other cysteine-rich proteins such as serum albumin.68 In addition, Au(I) thiolate compounds also interact with zinc finger proteins to trigger the release of zinc ions.69,70 In these examples, the ligands appear to play a role in stabilising or masking the reactive metal ion whilst transporting it through the body to the final target site, thus by altering the strength of the metal–ligand interactions the reactivity of the complex can be fine-tuned. This is a highly interesting strategy, but to the best of our knowledge there are no examples where the ligand has been designed to specifically direct the metal ion towards, and interact with, a specific enzyme.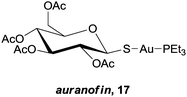
There are also a few examples of compounds where the ‘innocent’ ligand is designed to remain coordinated to the metal, and as seen with the inert complexes, the ligand can affect the selectivity of the complex. For example, the oxorhenium(V) complex 18 (Fig. 12) was designed to selectively inhibit the enzyme cathepsin B ahead of other members of the family, taking advantage of the occluding loop that extends over the active site of this enzyme. The tridentate sulphur ligand stabilises the Re(V) centre and the chloride ligand undergoes substitution with a thiol of the enzyme active site.71,72 The oxorhenium(V) complex is not only a nanomolar inhibitor of the enzyme, but is also 44-fold more selective for cathepsin B than cathepsin K (Fig. 12).
 | ||
| Fig. 12 The Re(V) complex 18 with cathepsin IC50 inhibition (μM) values. | ||
Active metal centres and active ligands
The concept of multi-targeted therapeutics to treat complex diseases, such as cancer, is gaining in popularity due to the ability of such a treatment regimens to attack the disease from multiple angles.73 Thus, by linking active metal centres to biologically active ligands, compounds capable of interacting with multiple biological targets in a semi-targeted fashion, can be accessed. Such hybrid complexes potentially offer advantages over drugs only capable of singular modes of action in that they are often more efficient and less vulnerable to the defence mechanisms of the disease. This approach, i.e. combining an organic enzyme inhibitor that interacts with the enzyme via non-covalent bonds, with a metal fragment that can directly coordinate to the enzyme, also leads to stronger inhibitors that are more likely to inhibit enzymes irreversibly. Such hybrid inhibitors offer several potential advantages including increased efficiency of target disruption, increased duration of action, and consequently less sensitivity toward pharmacokinetic parameters. However, if an inhibitor binds irreversibly to its target then it is important that the target is unique or significantly over-expressed in the diseased cells to avoid severe toxicity problems.The potential of this class of compound was demonstrated with rationally designed platinum compounds that target and inhibit glutathione-S-transferase (GST).74 GST's are an important class of enzymes which, among other things, are involved in the detoxification and removal of toxic compounds from the cell. In particular, GST P1-1 is implicated in drug resistance in some forms of cancer and is overexpressed in certain cisplatin-resistant tumours. Since reversal of this resistance can be achieved by inhibition of this enzyme, ethacraplatin, 20, which combines a Pt(IV) centre and a known GST inhibitor, ethacrynic acid (EA) 19, was developed (Fig. 13). Ethacraplatin was later shown to have a mode of action partly similar to satraplatin, i.e. involving in vivo reduction of Pt(IV) to the active Pt(II) centre, but in the case of ethacraplatin this step occurs inside the GST enzyme, with the two molecules of EA remaining within the enzyme, thus introducing a mechanism to overcome cisplatin resistance by the subsequent release of the Pt(II) ion. It was demonstrated that ethacraplatin is a low micromolar inhibitor of GST P1-1, more active than EA alone or in combination with cisplatin. In addition, 20 also retains cytotoxicity levels similar to cisplatin across a range of cell lines. A co-crystal structure of ethacraplatin with wild-type GST P1-1 revealed the enzyme is capable of activating and reducing the Pt centre, with both EA and the Pt ion initially bound to the enzyme (Fig. 14).75
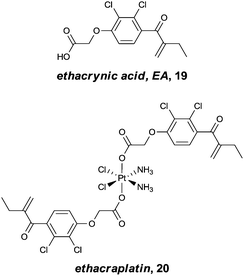 | ||
| Fig. 13 Ethacrynic acid (EA) and the ethacrynic acid–Pt(IV) complex, ethacraplatin, designed to overcome cisplatin resistance by inhibition of glutathione-S-transferase. | ||
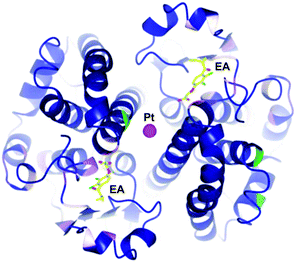 | ||
| Fig. 14 Structure of 20 bound to GST P1-1 showing cleavage of the Pt–EA moieties, with EA located in the enzyme active site, with the Pt at the dimer interface (PDB 3N9J).75 | ||
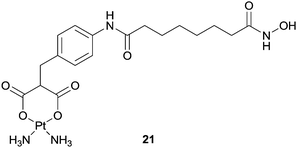
Using a similar approach, a SAHA–carboplatin conjugate, 21, was developed with the rationale that combining a platinum centre with an HDAC inhibitor could lead to a drug which is active against a broader spectrum of cancers, and also overcome drug resistance problems.76 In addition, SAHA has been shown to have some selectivity towards cancer cells over healthy cells, so the inclusion of such a moiety could also tackle the selectivity problems associated with current platinum based drugs. Carboplatin undergoes hydrolysis following cellular uptake, and it was proposed that the hybrid would act in a similar way, breaking apart in the nucleus to release the enzyme inhibitor and platinum unit which could exert their effects independently. Indeed, the compound is capable of binding to nucleotides and DNA in a similar manner to cisplatin, however, in a cell free assay the hybrid was approximately 100 fold less active than SAHA alone. In vitro cytotoxicity assays did not indicate any synergistic effects or improved activity against human ovarian cancer cell lines although selectivity towards non-tumour lines was observed.
One class of anti-cancer compounds which are promising with respect to being specifically toxic towards cancer cells are the ruthenium(II) arene compounds which contain a PTA ligand, 22, represented by the lead compound RAPTA-C 23 (Fig. 15).77,78 In contrast to Pt(II) drugs, RAPTA complexes favour interactions with proteins rather than DNA. For example, 23 has been shown to have a preference in the cell nucleus for histone protein sites as opposed to DNA within the nucleosome, as illustrated by the crystal structure of a ca. 200 kDa nucleosome core particle soaked with 23.79 With this in mind, and taking advantage of the robust nature of the RAPTA backbone, the prototype compound has been modified to tune the activity of the complexes.
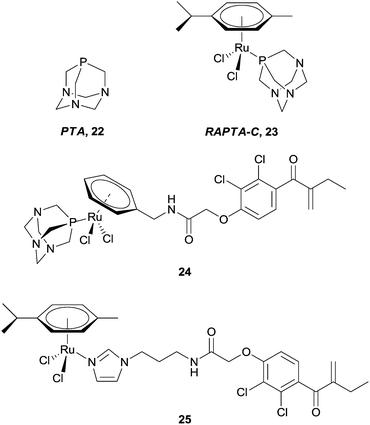 | ||
| Fig. 15 RAPTA-C and a selection of ruthenium(II) arene–EA conjugates. | ||
Applying the approach described for the platinum–ethacrynic acid derivative discussed above, the GST inhibitor EA was tethered to the ruthenium arene scaffold (Fig. 15) via the arene ring, e.g.24 (to retain the similarity with RAPTA complexes), or via an imadazole linker 25. Irrespective of the linking method, the compounds were found to be good inhibitors of GST, with IC50 values for the inhibition in the same order of magnitude as EA (12.0 μM), whereas the prototype compound, RAPTA-C, showed no inhibition of the enzyme up to 200 μM. In addition, the Ru–EA complexes were considerably more cytotoxic than RAPTA-C.80 Further experiments indicated that for the imidazole complexes (e.g.25), the molecule binds to a cysteine residue. A co-crystal structure of the RAPTA complex 24 with GST P1-1 indeed showed the metallodrug bound to the enzyme at the dimer interface, close to the thiol of Cys101. When the enzyme was treated with 24 for a longer time (48 h compared to 24 h), EA was found bound to the active site but the ruthenium was no longer present, consistent with mass spectrometric data which indicates dissociation of 24 over time. Such an observation could explain the increased activity of Ru–EA compared to RAPTA-C, as the ruthenium ion is released into a sensitised cell (Fig. 16).
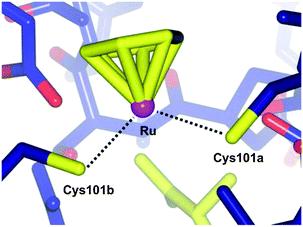 | ||
| Fig. 16 Diagram showing the bonding between the ruthenium centre of 24 and the two Cys101 residues of the individual monomers of GST P1-1 (PDB 3DD3).77 | ||
In attempts to address the problems associated with multi-drug resistance (MDR), Ru(II) arene complexes were combined with known inhibitors of the ATP-dependent efflux protein, P-glycoprotein (Pgp), which is often overexpressed in cancer cells. Furthermore, as efflux pumps are often non-specifically expressed in tumour cells, it was hoped that tethering MDR modulators to ruthenium compounds would overcome non-specificity, owing to the selective tumour-targeting properties of certain ruthenium compounds. Phenoxazine-based inhibitors were tethered to the ruthenium arene structure via an imidazole linker, e.g.27 (Fig. 17). In vitro assays revealed 27 retained selectivity towards tumorigenic cell lines and in addition, also inhibited Pgp, albeit less than verapamil (25%), an organic compound in clinical trials as a MDR antagonist. The imidazole ligand, phenoxide, 26, showed a comparable level of Pgp inhibition, but was not selective towards tumour cell lines indicating the ruthenium centre indeed confers some degree of selectivity.81
 | ||
| Fig. 17 A ruthenium(II) arene phenoxazine complex 27 designed as an MDR inhibitor and the uncoordinated ligand 26. | ||
Only recently, Nazarov and Dyson have reported on the conjugation of lonidamine, 28, to the ruthenium arene core (Fig. 18).82 Thus compounds such as 29 are expected to selectively interfere with cellular energy processes, as lonidamine is known to inhibit aerobic glycolysis in cancer cells whilst simultaneously enhancing aerobic glycolysis in normal, or healthy cells. Indeed, the hybrids show greater in vitro cytotoxicity (IC50ca. 20 μM) towards A2780 and A2780 cisplatin resistant cell lines than lonidamine (IC50ca. 200 μM), or a simple ruthenium–imidazole conjugate (IC50 > 100 μM). In addition, complex 29 shows moderate selectivity towards human glioblastoma cell lines relative to primary neuronal cultures of the cerebral cortex.
 | ||
| Fig. 18 Lonidamine 28, and an example of a ruthenium(II) arene–lonidamine hybrid 29 designed to inhibit cellular energy processes. | ||
In keeping with the theme of ruthenium(II) arene complexes, Salmain et al. described how complexes with chloroacetamide or maleimide groups tethered to the η6-bound arene ligand could direct the complex to the active site of the cysteine endoproteinase papain.83 Covalent binding of the complex to the enzyme was demonstrated by biochemical and mass spectrometric studies. Related iron-carbonyl derivatives were shown to behave in a related fashion,84 and other maleimide derivatives were shown to bind to thiol-containing biomolecules.85
Hartinger et al. have recently reported a series of complexes (30a–d) designed with a dual mode of action, exerting their effects by binding to DNA (via the ruthenium centre) whilst also inhibiting the enzyme topoisomerase II (via intercalation of the attached flavone ligand).86–88 In separate experiments, DNA binding and topoisomerase II inhibition (20–40 μM) was demonstrated, with the complexes acting as better inhibitors than the flavone ligands alone (>40 μM). Moreover, the cytotoxicity (low μM range) and topoisomerase inhibitory activity correlate well, suggesting their mechanism of action may involve inhibition of the enzyme. Although these are not the first examples of Ru-based topoisomerase inhibitors (or metal-based topoisomerase inhibitors), the authors were the first to demonstrate that the metal has an additional role to play in the mechanism by being able to interact with DNA. A series of ruthenium–naphthalimide conjugates 31 exert a related mechanism, i.e. the ruthenium centre interacts preferentially with proteins, leaving the naphthalimide unit free to intercalate to DNA. The enhanced cytotoxicity of the conjugates, compared with RAPTA-C, suggest an additional mechanism of action takes place, and on the basis of the intercalating behaviour of the naphthalimide unit, inhibition of topoisomerase cannot be discounted.89 A number of cytotoxic metal(II) arene complexes (metal = Ru and Os) with paullone ligands that exert their effect via the inhibition of kinases have also been reported (Fig. 19).90–92
 | ||
| Fig. 19 Examples of ruthenium(II) arene complexes with multiple modes of action. | ||
Lastly, and although their mode of action is unclear, non-steroidal anti-inflammatory drugs (NSAIDs), in particular aspirin 32, are being investigated as possible chemotherapeutic agents. It is believed they exert their effects via interruption of pathways associated with cyclooxygenase (COX) enzymes, especially COX-1 in the case of aspirin. However, the biological properties of the aspirin–hexacarbonyldicobalt conjugate 33 (Fig. 20)93 are significantly different from aspirin.34 Aspirin preferentially inhibits the enzyme COX-1 whereas 33 fails to discriminate between COX-1 and COX-2, which in turn is overexpressed in various tumours. In addition, 33 shows an activity profile similar to cisplatin across a range of cell lines.93,94 Although 33 has many sites susceptible to nucleophilic attack it is stable under in vitro conditions, suggesting the complex remains intact, and is responsible as a whole for the biological activity,95 probably via direct interaction with the enzyme and disruption of its expression. Further recent studies with zebra fish embryos also indicate that 33 also has anti-angiogenic properties, a trait which is not present with aspirin.96
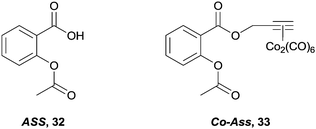 | ||
| Fig. 20 The indiscriminate COX-1 and COX-2 inhibitor, 33 and the parent organic compound, aspirin 32. | ||
Conclusions
The main ways in which metal complexes can inhibit enzymes have been categorized and illustrated with representative examples. Traditional metal-based drugs target DNA and simultaneously bind to many different proteins, but in this review we show that well designed metal complexes can exhibit higher degrees of selectivity relative to organic inhibitors and may also be more efficient inhibitors due to direct coordination. These complexes can be used as useful tools in chemical biology. However, since the emerging drugs used to treat diseases such as cancer are increasingly based on the inhibition of specific targets and pathways, there is clearly much promise for metal complexes that selectively inhibit enzymes in medicinal chemistry to treat a wide range of diseases. Future research may focus on non-toxic metals that are better detoxified and may have superior pharmacological profiles, especially when the metal is used as a scaffold. For active metals combined with organic inhibitors a more detailed mechanistic understanding of the role of coordination is required to establish the advantages and potential disadvantages of such systems at a molecular level. Approaches that allow the inclusion of structural diversity in a facile manner are also likely to become more widespread as this approach significantly increases the chances of discovering useful compounds.Acknowledgements
We thank the New Zealand Foundation of Research Science and Technology (Postdoctoral Fellowship, EPFL1001, to KJK) and the EPFL for funding.Notes and references
- B. Rosenberg, L. VanCamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS.
- T. Gianferrara, I. Bratsos and E. Alessio, Dalton Trans., 2009, 7588–7598 RSC.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817–7823 RSC.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS.
- S. P. Fricker, Dalton Trans., 2007, 4903–4917 RSC.
- E. Meggers, Angew. Chem., Int. Ed., 2011, 50, 2442–2448 CrossRef CAS.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS.
- P. J. Dyson and G. Sava, Dalton Trans., 2006, 1929–1933 RSC.
- G. Sava, A. Bergamo and P. J. Dyson, Dalton Trans., 2011, 40, 9069–9075 RSC.
- W. H. Ang and P. J. Dyson, Eur. J. Inorg. Chem., 2006, 3993 CrossRef.
- A. R. Timerbaev, C. G. Hartinger, S. S. Aleksenko and B. K. Keppler, Chem. Rev., 2006, 106, 2224–2248 CrossRef CAS.
- S. P. Mulcahy, S. Li, R. Korn, X. Xie and E. Meggers, Inorg. Chem., 2008, 47, 5030–5032 CrossRef CAS.
- J. X. Ong, C. W. Yap and W. H. Ang, Inorg. Chem., 2012, 51, 12483–12492 CrossRef CAS.
- C.-M. Che and F.-M. Siu, Curr. Opin. Chem. Biol., 2010, 14, 255–261 CrossRef CAS.
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS.
- A. Y. Louie and T. J. Meade, Chem. Rev., 1999, 99, 2711–2734 CrossRef CAS.
- E. Meggers, Chem. Commun., 2009, 1001–1010 RSC.
- G. Gasser and N. Metzler-Nolte, in Bioinorganic Medicinal Chemistry, e.d E. Alessio, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 351–382 Search PubMed.
- B. N. Trawick, A. T. Daniher and J. K. Bashkin, Chem. Rev., 1998, 98, 939–960 CrossRef CAS.
- J. Suh and W. S. Chei, Curr. Opin. Chem. Biol., 2008, 12, 207–213 CrossRef CAS.
- D. Salvemini, D. P. Riley and S. Cuzzocrea, Nat. Rev. Drug Discovery, 2002, 1, 367–374 CrossRef CAS.
- S. Top, J. Tang, A. Vessières, D. Carrez, C. Provot and G. Jaouen, Chem. Commun., 1996, 955–956 RSC.
- S. Top, B. Dauer, J. Vaissermann and G. Jaouen, J. Organomet. Chem., 1997, 541, 355–361 CrossRef CAS.
- A. Nguyen, A. Vessières, E. A. Hillard, S. Top, P. Pigeon and G. Jaouen, Chimia, 2007, 61, 716–724 CrossRef CAS.
- E. A. Hillard, A. Vessières and G. Jaouen, Top. Organomet. Chem., 2010, 32, 81–117 CrossRef CAS.
- E. Hillard, A. Vessières, L. Thouin, G. Jaouen and C. Amatore, Angew. Chem., Int. Ed., 2006, 45, 285–290 CrossRef CAS.
- C. Biot, G. Glorian, L. A. Maciejewski, J. S. Brocard, O. Domarle, G. Blampain, P. Millet, A. J. Georges, H. Abessolo, D. Dive and J. Lebibi, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS.
- A. Thiry, J.-M. Dogné, B. Masereel and C. T. Supuran, Trends Pharmacol. Sci., 2006, 27, 566–573 CrossRef CAS.
- S. Pastorekova, M. Zatovicova and J. Pastorek, Curr. Pharm. Des., 2008, 14, 685–698 CrossRef CAS.
- M. A. Glozak and E. Seto, Oncogene, 2007, 26, 5420–5432 CrossRef CAS.
- A. J. Salmon, M. L. Williams, A. Innocenti, D. Vullo, C. T. Supuran and S.-A. Poulsen, Bioorg. Med. Chem. Lett., 2007, 17, 5032–5035 CrossRef CAS.
- A. J. Salmon, M. L. Williams, A. Hofmann and S.-A. Poulsen, Chem. Commun., 2012, 48, 2328–2330 RSC.
- A. J. Salmon, M. L. Williams, Q. K. Wu, J. Morizzi, D. Gregg, S. A. Charman, D. Vullo, C. T. Supuran and S.-A. Poulsen, J. Med. Chem., 2012, 55, 5506–5517 CrossRef CAS.
- D. Can, B. Spingler, P. Schmutz, F. Mendes, P. Raposinho, C. Fernandes, F. Carta, A. Innocenti, I. Santos, C. T. Supuran and R. Alberto, Angew. Chem., Int. Ed., 2012, 51, 3354–3357 CrossRef CAS.
- F. W. Monnard, T. Heinisch, E. S. Nogueira, T. Schirmer and T. R. Ward, Chem. Commun., 2011, 47, 8238–8240 RSC.
- D. Can, H. W. Peindy N'Dongo, B. Spingler, P. Schmutz, P. Raposinho, I. Santos and R. Alberto, Chem. Biodiversity, 2012, 9, 1849–1866 CAS.
- J. Spencer, J. Amin, M. Wang, G. Packham, S. S. Syed Alwi, G. J. Tizzard, S. J. Coles, R. M. Paranal, J. E. Bradner and T. D. Heightman, ACS Med. Chem. Lett., 2011, 2, 358–362 CrossRef CAS.
- J. Spencer, J. Amin, R. Boddiboyena, G. Packham, B. E. Cavell, S. S. Syed Alwi, R. M. Paranal, T. D. Heightman, M. Wang, B. Marsden, P. Coxhead, M. Guille, G. J. Tizzard, S. J. Coles and J. E. Bradner, Med. Chem. Commun., 2012, 3, 61–64 RSC.
- E. Meggers, G. E. Atilla-Gokcumen, H. Bregman, J. Maksimoska, S. P. Mulcahy, N. Pagano and D. S. Williams, Synlett, 2007, 1177–1189 CrossRef CAS.
- J. É. Debreczeni, A. N. Bullock, G. E. Atilla, D. S. Williams, H. Bregman, S. Knapp and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 1580–1585 CrossRef CAS.
- J. Maksimoska, L. Feng, K. Harms, C. Yi, J. Kissil, R. Marmorstein and E. Meggers, J. Am. Chem. Soc., 2008, 130, 15764–15765 CrossRef CAS.
- G. E. Atilla-Gokcumen, N. Pagano, C. Streu, J. Maksimoska, P. Filippakopoulos, S. Knapp and E. Meggers, ChemBioChem, 2008, 9, 2933–2936 CrossRef CAS.
- P. Xie, D. S. Williams, G. E. Atilla-Gokcumen, L. Milk, M. Xiao, K. S. M. Smalley, M. Herlyn, E. Meggers and R. Marmorstein, ACS Chem. Biol., 2008, 3, 305–316 CrossRef CAS.
- L. Feng, Y. Geisselbrecht, S. Blanck, A. Wilbuer, G. E. Atilla-Gokcumen, P. Filippakopoulos, K. Kräling, M. A. Celik, K. Harms, J. Maksimoska, R. Marmorstein, G. Frenking, S. Knapp, L.-O. Essen and E. Meggers, J. Am. Chem. Soc., 2011, 133, 5976–5986 CrossRef CAS.
- S. Blanck, J. Maksimoska, J. Baumeister, K. Harms, R. Marmorstein and E. Meggers, Angew. Chem., Int. Ed., 2012, 51, 5244–5246 CrossRef CAS.
- D. S. Williams, P. J. Carroll and E. Meggers, Inorg. Chem., 2007, 46, 2944–2946 CrossRef CAS.
- J. Maksimoska, D. S. Williams, G. E. Atilla-Gokcumen, K. S. M. Smalley, P. J. Carroll, R. D. Webster, P. Filippakopoulos, S. Knapp, M. Herlyn and E. Meggers, Chem.–Eur. J., 2008, 14, 4816–4822 CrossRef.
- A. Wilbuer, D. H. Vlecken, D. J. Schmitz, K. Kräling, K. Harms, C. P. Bagowski and E. Meggers, Angew. Chem., Int. Ed., 2010, 49, 3839–3842 CrossRef CAS.
- S. Dieckmann, R. Riedel, K. Harms and E. Meggers, Eur. J. Inorg. Chem., 2012, 813–821 CrossRef CAS.
- S. Mollin, S. Blanck, K. Harms and E. Meggers, Inorg. Chim. Acta, 2012, 393, 261–268 CrossRef CAS.
- N. Pagano, J. Maksimoska, H. Bregman, D. S. Williams, R. D. Webster, F. Xue and E. Meggers, Org. Biomol. Chem., 2007, 5, 1218–1227 CAS.
- P. Xie, C. Streu, J. Qin, H. Bregman, N. Pagano, E. Meggers and R. Marmorstein, Biochemistry, 2009, 48, 5187–5198 CrossRef CAS.
- A. N. Bullock, S. Russo, A. Amos, N. Pagano, H. Bregman, J. É. Debreczeni, W. H. Lee, F. von Delft, E. Meggers and S. Knapp, PLoS One, 2009, 4, e7112 Search PubMed.
- G. Atilla-Gokcumen, L. Di Costanzo and E. Meggers, JBIC, J. Biol. Inorg. Chem., 2010, 16, 45–50 CrossRef.
- H. Bregman and E. Meggers, Org. Lett., 2006, 8, 5465–5468 CrossRef CAS.
- H. Bregman, P. J. Carroll and E. Meggers, J. Am. Chem. Soc., 2006, 128, 877–884 CrossRef CAS.
- A. Casini and J. Reedijk, Chem. Sci., 2012, 3, 3135–3144 RSC.
- A. Casini and L. Messori, Curr. Top. Med. Chem., 2011, 11, 2647–2660 CrossRef CAS.
- I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef CAS.
- S. J. Berners-Price and A. Filipovska, Metallomics, 2011, 3, 863–873 RSC.
- M. P. Rigobello, G. Scutari, A. Folda and A. Bindoli, Biochem. Pharmacol., 2004, 67, 689–696 CrossRef CAS.
- A. Chircorian and A. M. Barrios, Bioorg. Med. Chem. Lett., 2004, 14, 5113–5116 CrossRef CAS.
- C. E. W. K. Hashimoto, T. Matsubara, K. Hirohata and P. E. Lipsky, J. Clin. Invest., 1992, 89, 1839–1848 CrossRef.
- A. De Luca, C. G. Hartinger, P. J. Dyson, M. Lo Bello and A. Casini, J. Inorg. Biochem., 2013, 119, 38–42 CrossRef CAS.
- J. R. Roberts, J. Xiao, B. Schliesman, D. J. Parsons and C. F. Shaw, Inorg. Chem., 1996, 35, 424–433 CrossRef CAS.
- J. L. Larabee, J. R. Hocker and J. S. Hanas, Chem. Res. Toxicol., 2005, 18, 1943–1954 CrossRef CAS.
- F. Mendes, M. Groessl, A. A. Nazarov, Y. O. Tsybin, G. Sava, I. Santos, P. J. Dyson and A. Casini, J. Med. Chem., 2011, 54, 2196–2206 CrossRef CAS.
- R. Mosi, I. R. Baird, J. Cox, V. Anastassov, B. Cameron, R. T. Skerlj and S. P. Fricker, J. Med. Chem., 2006, 49, 5262–5272 CrossRef CAS.
- I. R. Baird, R. Mosi, M. Olsen, B. R. Cameron, S. P. Fricker and R. T. Skerlj, Inorg. Chim. Acta, 2006, 359, 2736–2750 CrossRef CAS.
- G. R. Zimmermann, J. Lehár and C. T. Keith, Drug Discovery Today, 2007, 12, 34–42 CrossRef CAS.
- W. H. Ang, I. Khalaila, C. S. Allardyce, L. Juillerat-Jeanneret and P. J. Dyson, J. Am. Chem. Soc., 2005, 127, 1382–1383 CrossRef CAS.
- L. J. Parker, L. C. Italiano, C. J. Morton, N. C. Hancock, D. B. Ascher, J. B. Aitken, H. H. Harris, P. Campomanes, U. Rothlisberger, A. De Luca, M. Lo Bello, W. H. Ang, P. J. Dyson and M. W. Parker, Chem.–Eur. J., 2011, 17, 7806–7816 CrossRef CAS.
- D. Griffith, M. P. Morgan and C. J. Marmion, Chem. Commun., 2009, 6735–6737 RSC.
- W. H. Ang, L. J. Parker, A. De Luca, L. Juillerat-Jeanneret, C. J. Morton, M. Lo Bello, M. W. Parker and P. J. Dyson, Angew. Chem., Int. Ed., 2009, 48, 3854–3857 CrossRef CAS.
- W. H. Ang, A. De Luca, C. Chapuis-Bernasconi, L. Juillerat-Jeanneret, M. Lo Bello and P. J. Dyson, ChemMedChem, 2007, 2, 1799–1806 CrossRef CAS.
- B. Wu, M. S. Ong, M. Groessl, Z. Adhireksan, C. G. Hartinger, P. J. Dyson and C. A. Davey, Chem.–Eur. J., 2011, 17, 3562–3566 CrossRef CAS.
- S. Chatterjee, I. Biondi, P. Dyson and A. Bhattacharyya, JBIC, J. Biol. Inorg. Chem., 2011, 16, 715–724 CrossRef CAS.
- C. A. Vock, W. H. Ang, C. Scolaro, A. D. Phillips, L. Lagopoulos, L. Juillerat-Jeanneret, G. Sava, R. Scopelliti and P. J. Dyson, J. Med. Chem., 2007, 50, 2166–2175 CrossRef CAS.
- A. A. Nazarov, D. Gardini, M. Baquié, L. Juillerat-Jeanneret, T. P. Serkova, E. P. Shevtsova, R. Scopelliti and P. Dyson, Dalton Trans., 2013, 42, 2347–2350 RSC.
- P. Haquette, B. Talbi, S. Canaguier, S. Dagorne, C. Fosse, A. Martel, G. Jaouen and M. Salmain, Tetrahedron Lett., 2008, 49, 4670–4673 CrossRef CAS.
- B. Rudolf, M. Salmain, E. Fornal and A. Rybarczyk-Pirek, Appl. Organomet. Chem., 2012, 26, 80–85 CrossRef CAS.
- M. Hanif, A. A. Nazarov, A. Legin, M. Groessl, V. B. Arion, M. A. Jakupec, Y. O. Tsybin, P. J. Dyson, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2012, 48, 1475–1477 RSC.
- A. Kurzwernhart, W. Kandioller, C. Bartel, S. Bächler, R. Trondl, G. Mühlgassner, M. A. Jakupec, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2012, 48, 4839–4841 RSC.
- A. Kurzwernhart, W. Kandioller, S. Bächler, C. Bartel, S. Martic, M. Buczkowska, G. Mühlgassner, M. A. Jakupec, H.-B. Kraatz, P. J. Bednarski, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, J. Med. Chem., 2012, 55, 10512–10522 CrossRef CAS.
- A. Kurzwernhart, W. Kandioller, E. A. Enyedy, M. Novak, M. A. Jakupec, B. K. Keppler and C. G. Hartinger, Dalton Trans., 2013 10.1039/c1032dt32206d.
- K. J. Kilpin, C. M. Clavel, F. Edafe and P. J. Dyson, Organometallics, 2012, 31, 7031–7039 CrossRef CAS.
- G. Mühlgassner, C. Bartel, W. F. Schmid, M. A. Jakupec, V. B. Arion and B. K. Keppler, J. Inorg. Biochem., 2012, 116, 180–187 CrossRef.
- I. N. Stepanenko, M. S. Novak, G. Mühlgassner, A. Roller, M. Hejl, V. B. Arion, M. A. Jakupec and B. K. Keppler, Inorg. Chem., 2011, 50, 11715–11728 CrossRef CAS.
- L. K. Filak, G. Mühlgassner, M. A. Jakupec, P. Heffeter, W. Berger, V. B. Arion and B. K. Keppler, JBIC, J. Biol. Inorg. Chem., 2010, 15, 903–918 CrossRef CAS.
- K. Schmidt, M. Jung, R. Keilitz, B. Schnurr and R. Gust, Inorg. Chim. Acta, 2000, 306, 6–16 CrossRef CAS.
- I. Ott, B. Kircher and R. Gust, J. Inorg. Biochem., 2004, 98, 485–489 CrossRef CAS.
- I. Ott and R. Gust, BioMetals, 2005, 18, 171–177 CrossRef CAS.
- I. Ott, B. Kircher, C. P. Bagowski, D. H. W. Vlecken, E. B. Ott, J. Will, K. Bensdorf, W. S. Sheldrick and R. Gust, Angew. Chem., Int. Ed., 2009, 48, 1160–1163 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |