 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Submillisecond-lived photoinduced charge separation in inclusion complexes composed of Li+@C60 and cyclic porphyrin dimers†
Takuya
Kamimura
a,
Kei
Ohkubo
b,
Yuki
Kawashima
b,
Hirofumi
Nobukuni
a,
Yoshinori
Naruta
a,
Fumito
Tani
*a and
Shunichi
Fukuzumi
*bc
aInstitute for Materials Chemistry and Engineering, Kyushu University, Fukuoka 812-8581, Japan. E-mail: tanif@ms.ifoc.kyushu-u.ac.jp; Fax: +81-92-642-2732; Tel: +81-92-642-2732
bDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA, Japan Science and Technology (JST), Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370; Tel: +81-6-6879-7368
cDepartment of Bioinspired Science, Ewha Womans University, Seoul, 120–750, Korea
First published on 8th January 2013
Abstract
Lithium ion encapsulated [60]fullerene (Li+@C60) is included within a free base and nickel complex of a cyclic porphyrin dimer (M-CPDPy, M = H4 and Ni2) to afford supramolecules (Li+@C60⊂M-CPDPy) in a polar solvent (benzonitrile) with the association constants of 2.6 × 105 M−1 and 3.5 × 105 M−1, respectively. From the electrochemical analysis, the energies of the charge-separated (CS) states are estimated to be 1.07 eV for Li+@C60⊂H4-CPDPy and 1.20 eV for Li+@C60⊂Ni2-CPDPy. Both values are lower than the triplet excited energies of the fullerene and porphyrin. Upon the photoexcitation at the Q-band of the porphyrin chromophore of Li+@C60⊂H4-CPDPy, electron transfer from the triplet excited state of the free base porphyrin to Li+@C60 occurs to produce the CS state. Li+@C60⊂Ni2-CPDPy also undergoes photoinduced electron transfer to produce the CS state. The lifetimes of the resulting CS states are 0.50 ms for Li+@C60⊂H4-CPDPy and 0.67 ms for Li+@C60⊂Ni2-CPDPy. These remarkably long CS lifetimes are the best values ever reported for non-covalent porphyrin-fullerene supramolecules in solution and are attributable to the lower CS energies than the triplet energy of each chromophore.
Introduction
In a natural photosynthetic reaction centre, the multistep electron-transfer reactions occur following the excitation of dimeric chlorophyll to attain the long-lived charge-separated (CS) state.1 The redox-active components such as chlorophyll, pheophytin and quinones are elegantly located in the protein matrix by non-covalent interactions.1 Extensive efforts have so far been devoted toward the design of electron donor–acceptor composites using covalently and non-covalently linked systems to form the long-lived CS state upon photoexcitation.2–26 The syntheses of covalently linked systems are inefficient and costly, thus it is a better choice to prepare supramolecular donor and acceptor systems.13–21Fullerenes and porphyrins are attractive building blocks for the construction of supramolecular electron donor–acceptor composites due to their excellent photophysical and electron-transfer properties.13–26 The fullerene derivatives have been widely employed as electron acceptors due to their favourable reduction potentials and small reorganization energy in electron-transfer reactions.20–28 On the other hand, the porphyrin compounds have very strong absorption bands in the visible region and their photoexcited states are generally good electron donors.28 Moreover, porphyrins are attractive components in the construction of host molecules for fullerenes through the π–π interactions between the spherical π-surface of fullerenes and flat π-planes of porphyrins.29–36 Numerous fullerene-porphyrin supramolecules as well as linked molecules have been extensively studied as functional models of the reaction centre for charge-separation process in natural photosynthesis.12–21,30–35 However, non-covalent binding between highly π-conjugated compounds such as porphyrins and fullerenes is not strong enough in polar solvents which are generally used for studies on photoinduced electron-transfer reactions. In contrast, when a non-polar solvent is used, the efficiency of charge separation is low in the supramolecular donor–acceptor complexes and the resulting CS state is extremely short-lived because the CS state decays to the triplet excited chromophore, which is lower in energy than the CS state.12,24b The energy of CS state should be lower than the triplet excited energy of each component. This is a typical dilemma for the long-lived charge separation in supramolecular donor–acceptor complexes.
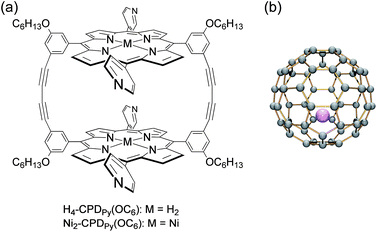
In order to solve this problem, we have recently reported the cyclic porphyrin dimers (CPDs) as shown in Scheme 1a.35 Receptor molecules composed of multiple porphyrins are suitable for the inclusion of pristine C60 and the derivatives. The strong supramolecular binding by π–π interaction was observed to form inclusion complexes.35,36 Unfortunately, the inclusion complex of C60 and the nickel cyclic porphyrin dimer (C60⊂Ni2-CPDPy) in crystalline state did not show the expected CS state in the time-resolved transient absorption spectra upon photoexcitation because the singlet excited state of the nickel porphyrin immediately gives rise to the triplet excited state by the rapid intersystem crossing, followed by energy transfer to afford the low-energy triplet excited state of C60 (3C60*).35 The estimated energy level of the CS state (1.98 eV) is higher than that of 3C60* (1.57 eV).35,37 In contrast, the corresponding inclusion complex of C60 and the free-base porphyrin dimer (C60⊂H4-CPDPy) underwent photoinduced electron transfer from the porphyrin to C60 owing to the lower oxidation potential and the slower intersystem crossing of the free-base porphyrin than those of the nickel complex. However, the lifetime of this CS state was very short probably because its energy level (1.83 eV) is still higher than that of 3C60*.35 The energy limit of a CS state is ca. 1.50–1.60 eV, which is the triplet excited energies of fullerenes and porphyrins.19
 | ||
| Scheme 1 Structures of (a) CPDPy(OC6) and (b) Li+@C60. | ||
It has been reported that a lithium ion encapsulated fullerene (Li+@C60) has a stronger electron accepting ability than pristine C60.38–42 The higher reduction potential of Li+@C60 than C60 makes the energy levels of the resulting CS states lower than the triplet excited energy.40,41 It is expected that the combination of Li+@C60 and the cyclic porphyrin dimers will make it possible to achieve both efficient formation of supramolecules and long-lived photoinduced charge separation. Thus, we report herein that supramolecular systems composed of cyclic porphyrin dimers and Li+@C60 afford photoinduced CS states with higher energies and longer lifetimes than those composed of Li+@C60 and monomeric porphyrins.41
Results and discussion
1. Supramolecular formation of cyclic porphyrin dimers with Li+@C60
Cyclic porphyrin dimers without OC6H13 groups have poor solubility in organic solvents.35,36 Therefore, we have designed and prepared new dimers with four long alkoxy substituents on the meso-phenyl groups (H4–, Ni2–CPDPy(OC6), Scheme 1) to improve the solubility. The synthetic procedures are shown in Scheme 2. The products were characterized by 1H-NMR, 13C-NMR, IR and mass spectroscopies (see the Experimental section and ESI† S1–S14). They are sufficiently soluble in benzonitrile (PhCN) to allow photochemical and electrochemical analysis with Li+@C60 in solution.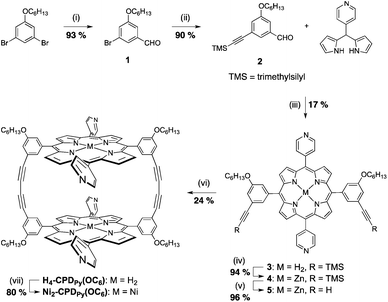 | ||
| Scheme 2 Synthetic pathway for H4–CPDPy(OC6) and Ni2–CPDPy(OC6). (i) n-Butyllithium, DMF, THF; (ii) trimethylsilylacetylene, Pd(OAc)2, Ph3P, Et3N; (iii) TFA, DDQ, CH2Cl2; (iv) Zn(OAc)2, CH2Cl2; (v) KF·2H2O, DMF/THF; (vi) CuCl, pyridine, air and (vii) Ni(OAc)2·4H2O, CHCl3/toluene (3/2 v/v). | ||
UV-vis absorption spectral changes were observed by addition of Li+@C60 PF6− to PhCN solutions of H4-CPDPy(OC6) and Ni2-CPDPy(OC6) as shown in Fig. 1 and Fig. S15 in ESI,† respectively. The Soret absorption bands of H4-CPDPy(OC6) and Ni2-CPDPy(OC6) were red-shifted and decreased in intensity with isosbestic points at 428 nm. The Job's plots upon mixing of Li+@C60 with H4-CPDPy(OC6) and Ni2-CPDPy(OC6) displayed typical signature patterns for the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest complex (Li+@C60⊂H4-CPDPy(OC6) and Li+@C60⊂Ni2-CPDPy(OC6), see Fig. S16 in ESI†). On the basis of the titration of H4-CPDPy(OC6) and Ni2-CPDPy(OC6) with Li+@C60 at 298 K, the association constants (Kassoc) were determined to be 2.6 × 105 M−1 for Li+@C60⊂H4-CPDPy(OC6) and 3.5 × 105 M−1 for Li+@C60⊂Ni2-CPDPy(OC6) by applying a nonlinear curve-fitting method using eqn (1) and (2),
:1 host–guest complex (Li+@C60⊂H4-CPDPy(OC6) and Li+@C60⊂Ni2-CPDPy(OC6), see Fig. S16 in ESI†). On the basis of the titration of H4-CPDPy(OC6) and Ni2-CPDPy(OC6) with Li+@C60 at 298 K, the association constants (Kassoc) were determined to be 2.6 × 105 M−1 for Li+@C60⊂H4-CPDPy(OC6) and 3.5 × 105 M−1 for Li+@C60⊂Ni2-CPDPy(OC6) by applying a nonlinear curve-fitting method using eqn (1) and (2),
| ΔAbs = L{1 + KassocA + KassocX − [(1 + KassocA + KassocX)2 − 4Kassoc2AX]0.5}/2KassocA | (1) |
| ΔInt = F{1 + KassocA + KassocX − [(1 + KassocA + KassocX)2 − 4Kassoc2AX]0.5}/2KassocA | (2) |
![(a) UV-vis absorption changes of H4-CPDPy(OC6) in the course of titration with Li+@C60 in PhCN at room temperature. [H4-CPDPy(OC6)] = 2.5 × 10−6 M. The inset shows the Soret band region. (b) Changes in the UV-vis absorbance (ΔAbs) at 422 nm. The curve was fitted by using eqn (1).](/image/article/2013/SC/c3sc22065f/c3sc22065f-f1.gif) | ||
| Fig. 1 (a) UV-vis absorption changes of H4-CPDPy(OC6) in the course of titration with Li+@C60 in PhCN at room temperature. [H4-CPDPy(OC6)] = 2.5 × 10−6 M. The inset shows the Soret band region. (b) Changes in the UV-vis absorbance (ΔAbs) at 422 nm. The curve was fitted by using eqn (1). | ||
Upon photoexcitation at the Soret band (430 nm) of H4–CPDPy(OC6) in PhCN, the fluorescence due to the porphyrin is observed at λmax = 650 and 717 nm as shown in Fig. 2. Addition of Li+@C60 to a PhCN solution of H4-CPDPy(OC6) induced a noticeable decrease in the fluorescence intensity of H4-CPDPy(OC6). From the plot of the fluorescence intensity change vs. the concentration of Li+@C60, the Kassoc value was determined to be 1.7 × 105 M−1, which agrees with the value obtained from the absorption spectral change within an experimental error (vide supra).
![(a) Fluorescence spectral changes of H4–CPDPy(OC6) in the course of titration with Li+@C60 in deaerated PhCN at 298 K excited at 430 nm. [H4–CPDPy(OC6)] = 2.5 × 10−6 M. (b) Changes in the fluorescence intensity (ΔInt) at 650 nm. The Kassoc value was evaluated by using eqn (2).](/image/article/2013/SC/c3sc22065f/c3sc22065f-f2.gif) | ||
| Fig. 2 (a) Fluorescence spectral changes of H4–CPDPy(OC6) in the course of titration with Li+@C60 in deaerated PhCN at 298 K excited at 430 nm. [H4–CPDPy(OC6)] = 2.5 × 10−6 M. (b) Changes in the fluorescence intensity (ΔInt) at 650 nm. The Kassoc value was evaluated by using eqn (2). | ||
2. Energetics of photoinduced processes
Cyclic voltammograms of Li+@C60⊂H4–CPDPy(OC6) and Li+@C60⊂Ni2–CPDPy(OC6) are shown in Fig. 3a and b. The comparison with the uncomplexed compounds shows that the cyclic voltammograms consist of the electron oxidation processes of CPDPy(OC6) and the electron reduction process of Li+@C60.43 The electrochemical data are summarized in Table 2. The energy of the CS states determined from the potential difference between one-electron reduction and oxidation potentials are 1.07 eV for Li+@C60⊂H4–CPDPy(OC6) and 1.20 eV for Li+@C60⊂Ni2–CPDPy(OC6). These values are smaller than those of the singlet excited states of cyclic porphyrin dimers (1.90 eV for H4–CPDPy(OC6),35c 1.97 eV for Ni2–CPDPy(OC6),35b and 1.94 eV for Li+@C60 (ref. 40)). Thus, the free energy changes of photoinduced electron transfer to Li+@C60via the singlet excited states are negative (exergonic).![Cyclic voltammograms (CV) and differential pulse voltammograms (DPV) of (a) Li+@C60⊂H4–CPDPy(OC6), (b) Li+@C60⊂Ni2–CPDPy(OC6), (c) C60⊂H4–CPDPy(OC6), and (d) C60⊂Ni2–CPDPy(OC6) in deaerated PhCN containing 0.10 M TBAPF6. (a and b) [CPDPy(OC6)] = [Li+@C60] = 2.5 × 10−4 M, (c and d) [CPDPy(OC6)] = [C60] = 2.0 × 10−4 M. Conditions: (a, b and d) 100 mV s−1 and (c) 500 mV s−1 for CV, and 4 mV s−1 for DPV.](/image/article/2013/SC/c3sc22065f/c3sc22065f-f3.gif) | ||
| Fig. 3 Cyclic voltammograms (CV) and differential pulse voltammograms (DPV) of (a) Li+@C60⊂H4–CPDPy(OC6), (b) Li+@C60⊂Ni2–CPDPy(OC6), (c) C60⊂H4–CPDPy(OC6), and (d) C60⊂Ni2–CPDPy(OC6) in deaerated PhCN containing 0.10 M TBAPF6. (a and b) [CPDPy(OC6)] = [Li+@C60] = 2.5 × 10−4 M, (c and d) [CPDPy(OC6)] = [C60] = 2.0 × 10−4 M. Conditions: (a, b and d) 100 mV s−1 and (c) 500 mV s−1 for CV, and 4 mV s−1 for DPV. | ||
| Compound | Reduction | Oxidation | CS energy, eV |
|---|---|---|---|
| E 1/2, V | E 1/2, V | ||
| Li+@C60⊂H4–CPDPy(OC6) | 0.14 | 1.21 | 1.07 |
| Li+@C60⊂Ni2–CPDPy(OC6) | 0.14 | 1.34 | 1.20 |
| C60⊂H4–CPDPy(OC6) | −0.46 | 1.40 | 1.86 |
| C60⊂Ni2–CPDPy(OC6) | −0.43 | 1.24 | 1.67 |
The energies of the triplet excited states were determined by phosphorescence spectra in a frozen PrCN/EtI (3:1 v/v) glasses at 77 K to be 1.51 eV for H4–CPDPy(OC6) and 1.50 eV for Ni2–CPDPy(OC6) (Fig. S18 in ESI†). The energy of the triplet excited state of Li+@C60 (3[Li+@C60]*; * denotes the excited state) was reported to be 1.53 eV.39,40 The energy level of the resulting CS states are lower than those of the triplet excited states of both Li+@C60 and the porphyrin dimers. Thus, photoinduced electron transfer from the singlet or triplet excited state of the porphyrin dimers to Li+@C60 as well as from the porphyrin dimers to the singlet or triplet excited state of Li+@C60 is energetically possible in the supramolecular complexes to form the CS states.
In contrast to the case of Li+@C60, the estimated energy levels of the CS states of C60⊂H4-CPDPy(OC6) and C60⊂Ni2-CPDPy(OC6) and (1.86 and 1.67 eV) are higher than those of the triplet excited state of C60 and CPDPy(OC6) as observed in our previous studies,35 suggesting no formation or short lifetimes of the CS states.
3. Photoinduced charge separation
The photodynamics of these inclusion complexes was investigated by the transient absorption spectra measured in PhCN by the use of femtosecond and nanosecond laser flash photolysis. The time-resolved transient absorption spectra of Li+@C60⊂H4–CPDPy(OC6) measured by femtosecond laser flash photolysis (λex = 420 nm) in the time range from 1 ps to 3000 ps (Fig. 4a), which showed little difference from the transient spectra of only H4–CPDPy(OC6) (Fig. S19a in ESI†), showing a characteristic absorption due to the singlet excited state of H4-CPDPy(OC6) (1[H4–CPDPy(OC6)]*). The decay rate constant of 1[H4–CPDPy(OC6)]* to the triplet excited state (3[H4–CPDPy(OC6)]*) was determined from the absorption change at 630 nm (inset of Fig. 4a) to be 1.0 × 109 s−1, which is slightly larger than the value of intersystem crossing of 1[H4-CPDPy(OC6)]* without Li+@C60 (kISC = 8.0 × 108 s−1) (Fig. S19a in ESI†). No characteristic absorption band due to Li+@C60˙− was observed at λmax = 1035 nm.39–41 Thus, no electron transfer from 1[H4–CPDPy(OC6)]* or 3[H4–CPDPy(OC6)]* to Li+@C60 occurred in the time range of femtosecond laser flash photolysis (∼3000 ps).![(a) Transient absorption spectra of H4–CPDPy(OC6) with Li+@C60 in deaerated PhCN at room temperature taken at 1, 100, and 3000 ps after femtosecond laser excitation at 420 nm. [H4–CPDPy(OC6)] = 7.0 × 10−6 M, [Li+@C60] = 1.4 × 10−5 M. (b) Transient absorption spectra of Ni2–CPDPy(OC6) with Li+@C60 in deaerated PhCN at room temperature taken at 1, 100, and 3000 ps after femtosecond laser excitation at 420 nm. [Ni2–CPDPy(OC6)] = 1.0 × 10−5 M, [Li+@C60] = 2.0 × 10−5 M.](/image/article/2013/SC/c3sc22065f/c3sc22065f-f4.gif) | ||
| Fig. 4 (a) Transient absorption spectra of H4–CPDPy(OC6) with Li+@C60 in deaerated PhCN at room temperature taken at 1, 100, and 3000 ps after femtosecond laser excitation at 420 nm. [H4–CPDPy(OC6)] = 7.0 × 10−6 M, [Li+@C60] = 1.4 × 10−5 M. (b) Transient absorption spectra of Ni2–CPDPy(OC6) with Li+@C60 in deaerated PhCN at room temperature taken at 1, 100, and 3000 ps after femtosecond laser excitation at 420 nm. [Ni2–CPDPy(OC6)] = 1.0 × 10−5 M, [Li+@C60] = 2.0 × 10−5 M. | ||
Similarly, the transient absorption spectra of Li+@C60⊂Ni2–CPDPy(OC6) measured by femtosecond transient absorption spectroscopy (Fig. 4b) indicates only the occurrence of the rapid intersystem crossing of 1[Ni2–CPDPy(OC6)]* to 3[Ni2–CPDPy(OC6)]* (kISC > 1012 s−1), in comparison with the transient spectra of only Ni2–CPDPy(OC6) (Fig. S19b in ESI†).44 The intersystem crossing process was not detected by use of our femtosecond laser system (fwhm = 130 fs). The kISC value to 3[Ni2–CPDPy(OC6)]* is much larger than that to 3[H4–CPDPy(OC6)]* because of the heavy atom effect of Ni. 3[N2–CPDPy(OC6)]* decayed to the ground state with the rate constant of kT = 5.3 × 109 s−1.
In contrast to the results of femtosecond laser flash photolysis in Fig. 4, where no electron transfer was observed, nanosecond laser excitation of Li+@C60⊂H4–CPDPy(OC6) resulted in observation of transient absorption bands at 690 and 1035 nm due to H4–CPDPy(OC6)˙+ and Li+@C60˙−, respectively (Fig. 5a).39–41 This clearly indicates the occurrence of electron transfer from 3[H4–CPDPy(OC6)]* to Li+@C60 to produce the CS state (Li+@C60˙−⊂H4–CPDPy(OC6)˙+). The rate of electron transfer from 3[H4–CPDPy(OC6)]* to Li+@C60 was too fast to detect in the time scale of the nanosecond laser flash photolysis experiments (kET > 107 s−1).45 The absorbance at 1035 nm due to Li+@C60˙− in the CS state decayed obeying first-order kinetics with the same slope irrespective of the difference in the laser intensity. This clearly indicates that the decay of the CS state occurs via intrasupramolecular back electron transfer rather than a bimolecular reaction. The CS lifetime was determined from the slope of the first-order plots in Fig. 5b to be 0.50 ms.
![(a) Transient absorption spectra of H4–CPDPy(OC6) with Li+@C60 in deaerated PhCN at room temperature taken at 30 and 200 μs after nanosecond laser excitation at 505 nm. [H4–CPDPy(OC6)] = 2.5 × 10−5 M, [Li+@C60] = 5.0 × 10−5 M. (b) Decay time profiles at 1035 nm with different laser intensities (1, 3, 5 mJ per pulse). Inset: first-order plots.](/image/article/2013/SC/c3sc22065f/c3sc22065f-f5.gif) | ||
| Fig. 5 (a) Transient absorption spectra of H4–CPDPy(OC6) with Li+@C60 in deaerated PhCN at room temperature taken at 30 and 200 μs after nanosecond laser excitation at 505 nm. [H4–CPDPy(OC6)] = 2.5 × 10−5 M, [Li+@C60] = 5.0 × 10−5 M. (b) Decay time profiles at 1035 nm with different laser intensities (1, 3, 5 mJ per pulse). Inset: first-order plots. | ||
Similarly nanosecond laser excitation of Li+@C60⊂Ni2–CPDPy(OC6) at 520 nm also results in formation of the CS state (Li+@C60˙−⊂Ni2–CPDPy(OC6)˙+) as shown in Fig. 6a, where transient absorption bands due to Ni2–CPDPy(OC6)˙+ and Li+@C60˙− were observed. In this case, however electron transfer occurs from Ni2–CPDPy(OC6) to the triplet excited state of Li+@C60 (3[Li+@C60]*) rather than from 3[Ni2–CPDPy(OC6)]* to Li+@C60 as indicated by the disappearance of the absorption band at 750 nm due to 3[Li+@C60]*, accompanied by the appearance of the absorption band at 1035 nm due to Li+@C60˙− (Fig. 6b). Photoexcitation at 520 nm where Li+@C60 has absorption results in the formation of 1[Li+@C60]*, which is converted to 3[Li+@C60]*via the intersystem crossing. The rate constant of electron transfer from Ni2–CPDPy(OC6) to 3[Li+@C60]* to produce the CS state was determined from the rise in the absorbance at 1035 nm due to Li+@C60˙− to be 5.7 × 107 s−1. The CS lifetime was determined from the slope of the first-order plots in Fig. 6c to be 0.67 ms, which is the longest value ever reported for non-covalent porphyrin-fullerene supramolecules in solution.41,46 The quantum yields of the CS states were estimated to be 0.32 for Li+@C60⊂H4–CPDPy(OC6) and 0.13 for Li+@C60⊂Ni2–CPDPy(OC6) by means of the comparative method with the absorption intensities of the CS states (Li+@C60˙−: ∑ (1035 nm) = 7300 M−1 cm−1).23,39
![(a) Transient absorption spectra of Ni2–CPDPy(OC6) with Li+@C60 in deaerated PhCN at room temperature taken at 4 and 28 μs after nanosecond laser excitation at 520 nm. [Ni2–CPDPy(OC6)] = 2.5 × 10−5 M, [Li+@C60] = 5.0 × 10−5 M. (b) Rise and (c) decay time profiles at 1035 nm with different laser intensities (1, 3, 5 mJ per pulse). Inset: first-order plots.](/image/article/2013/SC/c3sc22065f/c3sc22065f-f6.gif) | ||
| Fig. 6 (a) Transient absorption spectra of Ni2–CPDPy(OC6) with Li+@C60 in deaerated PhCN at room temperature taken at 4 and 28 μs after nanosecond laser excitation at 520 nm. [Ni2–CPDPy(OC6)] = 2.5 × 10−5 M, [Li+@C60] = 5.0 × 10−5 M. (b) Rise and (c) decay time profiles at 1035 nm with different laser intensities (1, 3, 5 mJ per pulse). Inset: first-order plots. | ||
When Li+@C60 was replaced by pristine C60, the transient absorption spectra of both C60⊂H4–CPDPy(OC6) and C60⊂Ni2–CPDPy(OC6) measured by nanosecond laser flash photolysis in PhCN exhibited only 740 nm bands for the triplet excited state of C60 with no transient absorption bands due to CPDPy(OC6)˙+ or C60˙− (Fig. S20 in ESI†). Thus, no CS states were produced as predicted by their higher energy levels than those of the triplet excited states of CPDPy(OC6) and C60.
The temperature dependence of the charge recombination (CR) process was investigated by nanosecond laser flash photolysis in the range of 25–70 °C for Li+@C60⊂H4–CPDPy(OC6) and 25–80 °C for Li+@C60⊂Ni2–CPDPy(OC6). The activation enthalpies were determined from the Eyring plots of the rate constants (kBET) of the back electron transfer (charge recombination) to be 3.5 kcal mol−1 for both Li+@C60⊂H4–CPDPy(OC6) and Li+@C60⊂Ni2–CPDPy(OC6) (Fig. S21 in ESI†).
The linear plots of ln(kBETT1/2) vs. T−1 derived from the Marcus equation (eqn (3))47 in Fig. 7 afford the reorganization
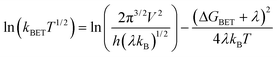 | (3) |
![Plots of ln(kBETT1/2) vs. T−1 for charge recombination of [H4–CPDPy(OC6)˙+ + Li+@C60˙−] (black) and [Ni2–CPDPy(OC6)˙+ + Li+@C60˙−] (red) in PhCN.](/image/article/2013/SC/c3sc22065f/c3sc22065f-f7.gif) | ||
| Fig. 7 Plots of ln(kBETT1/2) vs. T−1 for charge recombination of [H4–CPDPy(OC6)˙+ + Li+@C60˙−] (black) and [Ni2–CPDPy(OC6)˙+ + Li+@C60˙−] (red) in PhCN. | ||
Based on the results described above, the mechanisms of intrasupramolecular photoinduced charge separation in Li+@C60⊂H4–CPDPy(OC6) and Li+@C60⊂Ni2–CPDPy(OC6) are proposed as shown in Schemes 3 and 4, respectively. The singlet excited state of H4–CPDPy(OC6) (1[H4–CPDPy(OC6)]*) is generated upon photoexcitation of Li+@C60⊂H4–CPDPy(OC6) at 420 nm, where the porphyrin moiety is exclusively excited. Even if the Li+@C60 moiety is excited, energy transfer from 1[Li+@C60]* (Es = 1.94 eV) to H4–CPDPy(OC6) (Es = 1.90 eV) may occur to produce 1[H4–CPDPy(OC6)]*. Although electron transfer from 1[H4–CPDPy(OC6)]* to Li+@C60 energetically possible (Scheme 3), the intersystem crossing to generate 3[H4–CPDPy(OC6)]* occurs with the rate constant of 8.3 × 108 s−1. Then, electron transfer occurs from 3[H4–CPDPy(OC6)]* to Li+@C60 with the driving force of 0.44 eV to produce the CS state with a much larger rate constant (kET > 107 s−1) than the triplet decay to the ground state (kT = 2.1 × 103 s−1). The CS state decays slowly via intrasupramolecular BET with the lifetime of 0.50 ms (Scheme 3).
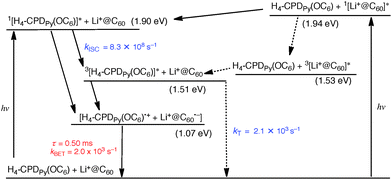 | ||
| Scheme 3 Energy diagram for Li+@C60⊂H4–CPDPy(OC6); broken arrow: minor pathway. | ||
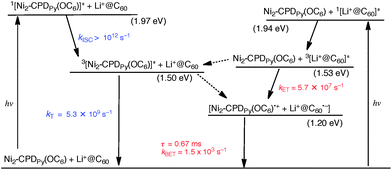 | ||
| Scheme 4 Energy diagram for Li+@C60⊂Ni2–CPDPy(OC6); broken arrow: minor pathway. | ||
As the case of Li+@C60⊂Ni2–CPDPy(OC6), photoexcitation of Li+@C60⊂Ni2–CPDPy(OC6) at 420 nm, where the porphyrin moiety is excited exclusively, results in formation of the singlet excited state (1[Ni2–CPDPy(OC6)]*) as shown in Scheme 4. In contrast to the case of H4–CPDPy(OC6), however, the intersystem crossing to 3[Ni2–CPDPy(OC6)]* occurs rapidly with the rate constant (kISC > 1012 s−1). 3[Ni2–CPDPy(OC6)]* decays to the ground state prior to electron transfer from 3[Ni2–CPDPy(OC6)]* to Li+@C60.48 Photoexcitation of Li+@C60⊂Ni2–CPDPy(OC6) at 520 nm, where Li+@C60 has absorption, generates the singlet excited state (1[Li+@C60]*). After the intersystem crossing, electron transfer from Ni2–CPDPy(OC6) to 3[Li+@C60]* occurs with the rate constant of 5.7 × 107 s−1 to produce the CS state. The CS state decays slowly with the lifetime of 0.67 ms (Scheme 4).
Conclusions
Li+@C60 is included within a free base and nickel complex of a cyclic porphyrin dimer to give stable supramolecules in benzonitrile. From the electrochemical analysis, the energies of the expected CS states are estimated to be lower than the triplet excited energies of the fullerene and porphyrin. Li+@C60⊂H4–CPDPy(OC6) undergoes photoinduced electron transfer from the triplet excited state of the porphyrin to Li+@C60 to afford the CS state. Li+@C60⊂Ni2–CPDPy(OC6) also undergoes photoinduced electron transfer from the nickel porphyrin to the triplet excited state of Li+@C60 with a rate constant of 5.7 × 107 s−1. The lifetimes of the resulting CS states are 0.50 ms for Li+@C60⊂H4–CPDPy(OC6) and 0.67 ms for Li+@C60⊂Ni2–CPDPy(OC6). These CS lifetimes are the longest values ever reported for non-covalent porphyrin-fullerene supramolecules in solution and are attributed to the lower CS energies than the triplet energy of each chromophore.Experimental section
Materials
Reagents and solvents of best grade available were purchased from commercial suppliers and were used without further purification unless otherwise noted. Lithium ion-encapsulated C60 (Li+@C60 PF6−: 96%) was obtained from Daiichi Jitsugyo Co. Ltd, Japan. N,N-Dimethylformamide (DMF) was purified by distillation from CaH2 under reduced pressure. Benzonitrile (PhCN) was purified by distillation from P2O5 under reduced pressure after being stirred with K2CO3 overnight. Dry tetrahydrofuran (THF) was obtained by distillation from Na and benzophenone under N2 atmosphere. Dry triethylamine (Et3N) was obtained by distillation from CaH2 under N2 atmosphere, after being stirred with KOH overnight.NMR and mass measurements
Nuclear magnetic resonance (NMR) spectra were recorded on a JEOL JNM-ECS400 (400 MHz for 1H), JEOL JNM-ECA500 (500 MHz for 1H), or Bruker AVANCE III 600 (151 MHz for 13C) spectrometer. Chemical shifts were reported as δ values in ppm relative to tetramethylsilane. High-resolution fast atom bombardment mass spectra (HR-FAB-MS) were measured with 3-nitrobenzyl alcohol (NBA) as a matrix and recorded on a JEOL LMS-HX-110 spectrometer.UV-vis and IR absorption measurements
Ultraviolet-visible (UV-vis) absorption and infrared (IR) spectra were recorded on Shimadzu UV-3100PC and BIO RAD FTS6000 spectrophotometers, respectively.Emission measurements
Fluorescence spectra were measured on a Horiba FluoroMax-4 spectrofluorophotometer with a quartz cuvette (path length = 10 mm) at 298 K. Phosphorescence spectra were measured on a Horiba Fluorolog τ3 spectrophotometer with a quartz tube (i.d. = 4 mm) at 77 K.Electrochemical measurements
Electrochemical measurements were performed on a ALS630B electrochemical analyzer in deaerated PhCN containing 0.1 M Bu4NPF6 as the supporting electrolyte at 298 K. A conventional three-electrode cell was used with a platinum working electrode (surface area of 0.3 mm2) and a platinum wire as a counter electrode. The platinum working electrodes (BAS) were routinely polished with BAS polishing alumina suspension and rinsed with acetone and acetonitrile before use. The measured potentials were recorded with respect to an Ag/AgNO3 (0.01 M) reference electrode. All potentials (vs. Ag/Ag+) were converted to values vs. SCE by adding 0.29 V. All electrochemical measurements were carried out under an N2 atmosphere.Laser flash photolysis
Femtosecond transient absorption spectroscopy experiments were conducted using an ultrafast source: Integra-C (Quantronix Corp.), an optical parametric amplifier: TOPAS (Light Conversion Ltd) and a commercially available optical detection system: Helios provided by Ultrafast Systems LLC. The source for the pump and probe pulses were derived from the fundamental output of Integra-C (λ = 786 nm, 2 mJ per pulse and fwhm = 130 fs) at a repetition rate of 1 kHz. 75% of the fundamental output of the laser was introduced into a second harmonic generation (SHG) unit: Apollo (Ultrafast Systems) for excitation light generation at λ = 393 nm, while the rest of the output was used for white light generation. The laser pulse was focused on a sapphire plate of 3 mm thickness and then white light continuum covering the visible region from λ = 410 nm to 800 nm was generated via self-phase modulation. A variable neutral density filter, an optical aperture, and a pair of polarizer were inserted in the path in order to generate stable white light continuum. Prior to generating the probe continuum, the laser pulse was fed to a delay line that provides an experimental time window of 3.2 ns with a maximum step resolution of 7 fs. In our experiments, a wavelength at λ = 393 nm of SHG output was irradiated at the sample cell with a spot size of 1 mm diameter where it was merged with the white probe pulse in a close angle (<10°). The probe beam after passing through the 2 mm sample cell was focused on a fiber optic cable that was connected to a CMOS spectrograph for recording the time-resolved spectra (λ = 410–800 nm). Typically, 1500 excitation pulses were averaged for 3 seconds to obtain the transient spectrum at a set delay time. Kinetic traces at appropriate wavelengths were assembled from the time-resolved spectral data. All measurements were conducted at room temperature, 295 K.Nanosecond time-resolved transient absorption measurements were carried out using the laser system provided by UNISOKU Co., Ltd. Measurements of nanosecond transient absorption spectrum were performed according to the following procedure. A deaerated solution containing supramolecule was excited by a Panther OPO pumped by a Nd:YAG laser (Continuum SLII-10, 4–6 ns fwhm). The photodynamics were monitored by continuous exposure to a xenon lamp (150 W) as a probe light and a photomultiplier tube (Hamamatsu 2949) as a detector. The solution was oxygenated by nitrogen purging for 15 min prior to measurements.
Synthesis
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 103.5 (CC), 113.9 (aromatic C), 124.3 (aromatic C), 125.3 (aromatic C), 126.9 (aromatic C), 137.8 (aromatic C), 159.6 (aromatic C), 191.6 (CHO); HR-FAB-MS (NBA): m/z calcd for C18H27O2Si: 303.1780; found: 303.1789; elemental analysis (%) calcd for C18H26O2Si: C 71.47 H 8.66; found: C 71.49 H 8.74.
C), 105.1 (CC), 117.2 (aromatic), 117.3 (aromatic), 119.9 (aromatic C), 122.5 (aromatic C), 129.5 (aromatic C), 130.8 (aromatic C), 143.1 (aromatic C), 148.5 (aromatic C), 150.3 (aromatic C), 157.5 (aromatic C); HR-FAB-MS (NBA): m/z calcd for C64H68N6O2Si2: 1008.4942; found: 1008.4897; elemental analysis (%) calcd for C64H68N6O2Si2: C 76.15 H 6.79 N 8.33; found: C 76.05 H 6.80 N 8.12; UV-vis (CHCl3): λmax (ε, cm−1 M−1) 420 (447800), 515 (19500), 549 (5700), 588 (5900), 646 (2700); IR (KBr): ν = 3318, 2955, 2930, 2870, 2158, 1591, 1475, 1421, 1354, 1249, 1159, 976, 928, 855, 801, 760, 730, 660 cm−1.
C), 106.3 (CC), 117.2 (aromatic C), 117.2 (aromatic C), 118.7 (aromatic C), 120.7 (aromatic C), 122.7 (aromatic C), 130.1 (aromatic C), 131.5 (aromatic C), 132.1 (aromatic C), 132.8 (aromatic C), 145.2 (aromatic C), 148.7 (aromatic C), 149.8 (aromatic C), 150.7 (aromatic C), 151.4 (aromatic C), 157.9 (aromatic C); HR-FAB-MS (NBA): m/z calcd for C64H66N6O2Si2Zn: 1070.4077; found: 1070.4080; elemental analysis (%) calcd for C64H66N6O2Si2Zn·0.5CH4O: C 71.15 H 6.30 N 7.72; found: C 71.20 H 6.21 N 7.67; UV-vis (CHCl3): λmax (ε, cm−1 M−1) 425 (455800), 554 (18000), 595 (3500); IR (KBr): ν = 2955, 2931, 2157, 1592, 1419, 1346, 1249, 1161, 1072, 998, 945, 855, 795, 760, 718, 667 cm−1.
CH), 7.77 (br-s, 2H, Ar–H), 8.15 (d, J = 6.3 Hz, 2H, Ar–H), 8.30–8.33 (m, 6H, Ar–H), 9.10 (d, J = 4.6 Hz, 4H, pyrrole β-H), 9.18 (d, J = 5.2 Hz, 4H, Ar–H), 9.26 (d, J = 4.6 Hz, 4H, pyrrole β-H); 13C NMR (pyridine-d5, 151 MHz): δ 14.2 (CH3), 22.8 (CH2), 26.0 (CH2), 29.6 (CH2), 31.8 (CH2), 68.7(OCH2), 79.6 (CCH), 84.6 (CCH), 117.5 (aromatic C), 117.6 (aromatic C), 118.8 (aromatic C), 120.7 (aromatic C), 122.0 (aromatic C), 130.1 (aromatic C), 131.6 (aromatic C), 132.2 (aromatic C), 132.9 (aromatic C), 145.3 (aromatic C), 148.7 (aromatic C), 149.8 (aromatic C), 150.7 (aromatic C), 151.4 (aromatic C), 157.9 (aromatic C); HR-FAB-MS (NBA): m/z calcd for C58H50N6O2Zn: 926.3287; found: 926.3279; elemental analysis (%) calcd for C58H50N6O2Zn·0.5CH4O: C 74.39 H 5.55 N 8.90; found: C 74.33 H 5.36 N 8.84; UV-vis (CHCl3): λmax (ε, cm−1 M−1) 425 (463000), 553 (18300), 594 (3500); IR (KBr): ν = 2952, 2929, 2869, 1592, 1419, 1344, 1322, 1204, 1180, 1144, 1072, 998, 938, 795, 718, 676 cm−1.
C), 83.2 (CC), 114.9 (aromatic C), 117.1 (aromatic C), 118.8 (aromatic C), 120.2 (aromatic C), 120.8 (aromatic C), 129.3 (aromatic C), 135.0 (aromatic C), 143.0 (aromatic C), 148.4 (aromatic C), 149.9 (aromatic C), 158.0 (aromatic C); HR-FAB-MS (NBA): m/z calcd for C116H100N12O4: 1724.7991; found: 1724.7997; elemental analysis (%) calcd for C116H100N12O4·C3H8O: C 80.01 H 6.09 N 9.41; found: C 80.12 H 5.90 N 9.44; UV-vis (CHCl3): λmax (ε, cm−1 M−1) 419 (845300), 515 (36100), 549 (9200), 589 (10800), 646 (4300); IR (KBr): ν = 2953, 2928, 2869, 1590, 1475, 1417, 1370, 1195, 1171, 1056, 976, 799, 728, 660 cm−1.
C), 81.8 (CC), 115.8 (aromatic C), 115.9 (aromatic C), 117.1 (aromatic C), 120.6 (aromatic C), 121.1 (aromatic C), 128.5 (aromatic C), 132.1 (aromatic C), 132.2 (aromatic C), 133.0 (aromatic C), 141.11 (aromatic C), 141.5 (aromatic C), 141.9 (aromatic C), 148.6 (aromatic C), 148.7 (aromatic C), 158.2 (aromatic C); HR-FAB-MS (NBA): m/z calcd for C116H96N12O4Ni2: 1836.6384; found: 1836.6384; elemental analysis (%) calcd for C116H96N12O4Ni2·0.5C3H8O: C 75.48 H 5.39 N 8.99; found: C 75.30 H 5.11 N 9.02; UV-vis (CHCl3): λmax (ε, cm−1 M−1) 415 (464000), 532 (30000); IR (KBr): ν = 2952, 2928, 2869, 1593, 1418, 1372, 1182, 1009, 796, 713, 671 cm−1.
C), 103.5 (CC), 113.9 (aromatic C), 124.3 (aromatic C), 125.3 (aromatic C), 126.9 (aromatic C), 137.8 (aromatic C), 159.6 (aromatic C), 191.6 (CHO); HR-FAB-MS (NBA): m/z calcd for C18H27O2Si: 303.1780; found: 303.1789; elemental analysis (%) calcd for C18H26O2Si: C 71.47 H 8.66; found: C 71.49 H 8.74.
C), 105.1 (CC), 117.2 (aromatic), 117.3 (aromatic), 119.9 (aromatic C), 122.5 (aromatic C), 129.5 (aromatic C), 130.8 (aromatic C), 143.1 (aromatic C), 148.5 (aromatic C), 150.3 (aromatic C), 157.5 (aromatic C); HR-FAB-MS (NBA): m/z calcd for C64H68N6O2Si2: 1008.4942; found: 1008.4897; elemental analysis (%) calcd for C64H68N6O2Si2: C 76.15 H 6.79 N 8.33; found: C 76.05 H 6.80 N 8.12; UV-vis (CHCl3): λmax (ε, cm−1 M−1) 420 (447800), 515 (19500), 549 (5700), 588 (5900), 646 (2700); IR (KBr): ν = 3318, 2955, 2930, 2870, 2158, 1591, 1475, 1421, 1354, 1249, 1159, 976, 928, 855, 801, 760, 730, 660 cm−1.
C), 106.3 (CC), 117.2 (aromatic C), 117.2 (aromatic C), 118.7 (aromatic C), 120.7 (aromatic C), 122.7 (aromatic C), 130.1 (aromatic C), 131.5 (aromatic C), 132.1 (aromatic C), 132.8 (aromatic C), 145.2 (aromatic C), 148.7 (aromatic C), 149.8 (aromatic C), 150.7 (aromatic C), 151.4 (aromatic C), 157.9 (aromatic C); HR-FAB-MS (NBA): m/z calcd for C64H66N6O2Si2Zn: 1070.4077; found: 1070.4080; elemental analysis (%) calcd for C64H66N6O2Si2Zn·0.5CH4O: C 71.15 H 6.30 N 7.72; found: C 71.20 H 6.21 N 7.67; UV-vis (CHCl3): λmax (ε, cm−1 M−1) 425 (455800), 554 (18000), 595 (3500); IR (KBr): ν = 2955, 2931, 2157, 1592, 1419, 1346, 1249, 1161, 1072, 998, 945, 855, 795, 760, 718, 667 cm−1.
CH), 7.77 (br-s, 2H, Ar–H), 8.15 (d, J = 6.3 Hz, 2H, Ar–H), 8.30–8.33 (m, 6H, Ar–H), 9.10 (d, J = 4.6 Hz, 4H, pyrrole β-H), 9.18 (d, J = 5.2 Hz, 4H, Ar–H), 9.26 (d, J = 4.6 Hz, 4H, pyrrole β-H); 13C NMR (pyridine-d5, 151 MHz): δ 14.2 (CH3), 22.8 (CH2), 26.0 (CH2), 29.6 (CH2), 31.8 (CH2), 68.7(OCH2), 79.6 (CCH), 84.6 (CCH), 117.5 (aromatic C), 117.6 (aromatic C), 118.8 (aromatic C), 120.7 (aromatic C), 122.0 (aromatic C), 130.1 (aromatic C), 131.6 (aromatic C), 132.2 (aromatic C), 132.9 (aromatic C), 145.3 (aromatic C), 148.7 (aromatic C), 149.8 (aromatic C), 150.7 (aromatic C), 151.4 (aromatic C), 157.9 (aromatic C); HR-FAB-MS (NBA): m/z calcd for C58H50N6O2Zn: 926.3287; found: 926.3279; elemental analysis (%) calcd for C58H50N6O2Zn·0.5CH4O: C 74.39 H 5.55 N 8.90; found: C 74.33 H 5.36 N 8.84; UV-vis (CHCl3): λmax (ε, cm−1 M−1) 425 (463000), 553 (18300), 594 (3500); IR (KBr): ν = 2952, 2929, 2869, 1592, 1419, 1344, 1322, 1204, 1180, 1144, 1072, 998, 938, 795, 718, 676 cm−1.
C), 83.2 (CC), 114.9 (aromatic C), 117.1 (aromatic C), 118.8 (aromatic C), 120.2 (aromatic C), 120.8 (aromatic C), 129.3 (aromatic C), 135.0 (aromatic C), 143.0 (aromatic C), 148.4 (aromatic C), 149.9 (aromatic C), 158.0 (aromatic C); HR-FAB-MS (NBA): m/z calcd for C116H100N12O4: 1724.7991; found: 1724.7997; elemental analysis (%) calcd for C116H100N12O4·C3H8O: C 80.01 H 6.09 N 9.41; found: C 80.12 H 5.90 N 9.44; UV-vis (CHCl3): λmax (ε, cm−1 M−1) 419 (845300), 515 (36100), 549 (9200), 589 (10800), 646 (4300); IR (KBr): ν = 2953, 2928, 2869, 1590, 1475, 1417, 1370, 1195, 1171, 1056, 976, 799, 728, 660 cm−1.
C), 81.8 (CC), 115.8 (aromatic C), 115.9 (aromatic C), 117.1 (aromatic C), 120.6 (aromatic C), 121.1 (aromatic C), 128.5 (aromatic C), 132.1 (aromatic C), 132.2 (aromatic C), 133.0 (aromatic C), 141.11 (aromatic C), 141.5 (aromatic C), 141.9 (aromatic C), 148.6 (aromatic C), 148.7 (aromatic C), 158.2 (aromatic C); HR-FAB-MS (NBA): m/z calcd for C116H96N12O4Ni2: 1836.6384; found: 1836.6384; elemental analysis (%) calcd for C116H96N12O4Ni2·0.5C3H8O: C 75.48 H 5.39 N 8.99; found: C 75.30 H 5.11 N 9.02; UV-vis (CHCl3): λmax (ε, cm−1 M−1) 415 (464000), 532 (30000); IR (KBr): ν = 2952, 2928, 2869, 1593, 1418, 1372, 1182, 1009, 796, 713, 671 cm−1.
Acknowledgements
This work was supported by Grants-in-Aid (Scientific Research on Innovative Areas nos. 20108010 to S.F. and 20108009 to F.T., “pi-Space”, 23750014 to K.O., and KOSEF/MEST through WCU project (R31-2008-000-10010-0), Korea. F.T. acknowledges Tokuyama Science Foundation and Iketani Science and Technology Foundation for Research Grants.Notes and references
- (a) A. J. Hoff and J. Deisenhofer, Phys. Rep., 1997, 287, 1 CrossRef CAS; (b) Anoxygenic Photosynthetic Bacteria, ed. R. E. Blankenship, M. T. Madigan and C. E. Bauer, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1995 Search PubMed.
- (a) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS; (b) G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768 CrossRef CAS; (c) D. Gust and T. A. Moore, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, CA, 2000, vol. 8, p. 153 Search PubMed; (d) D. Gust, T. A. Moore and A. L. Moore, in Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, vol. 3, p. 272 Search PubMed.
- (a) D. M. Guldi, G. M. A. Rahman, F. Zerbetto and M. Prato, Acc. Chem. Res., 2005, 38, 871 CrossRef CAS; (b) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS; (c) V. Sgobba and D. M. Guldi, Chem. Soc. Rev., 2009, 38, 165 RSC.
- (a) M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS; (b) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS.
- (a) N. V. Tkachenko, L. Rantala, A. Y. Tauber, J. Helaja, P. H. Hynninen and H. Lemmetyinen, J. Am. Chem. Soc., 1999, 121, 9378 CrossRef CAS; (b) M. Isosomppi, N. V. Tkachenko, A. Efimov and H. Lemmetyinen, J. Phys. Chem. A, 2005, 109, 4881 CrossRef CAS; (c) S. A. Vail, D. I. Schuster, D. M. Guldi, M. Isosomppi, N. Tkachenko, H. Lemmetyinen, A. Palkar, L. Echegoyen, X. Chen and J. Z. H. Zhang, J. Phys. Chem. B, 2006, 110, 14155 CrossRef CAS; (d) D. I. Schuster, K. Li, D. M. Guldi, A. Palkar, L. Echegoyen, C. Stanisky, R. J. Cross, M. Niemi, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2007, 129, 15973 CrossRef CAS.
- J. L. Sessler, B. Wang, S. L. Springs and C. T. Brown, in Comprehensive Supramolecular Chemistry, ed. J. L. Atwood, J. E. D. Davies, D. D. MacNicol and F. Vögtle, Elsevier Science Ltd, Oxford, 1999, vol. 4, p. 311 Search PubMed.
- A. Osuka, N. Mataga and T. Okada, Pure Appl. Chem., 1997, 69, 797 CrossRef CAS.
- (a) K. D. Jordan and M. N. Paddon-Row, Chem. Rev., 1992, 92, 395 CrossRef CAS; (b) M. N. Paddon-Row, Acc. Chem. Res., 1994, 27, 18 CrossRef CAS.
- M.-S. Choi, T. Yamazaki, I. Yamazaki and T. Aida, Angew. Chem., Int. Ed., 2004, 43, 150 CrossRef CAS.
- (a) S. Fukuzumi, Org. Biomol. Chem., 2003, 1, 609 RSC; (b) S. Fukuzumi, Bull. Chem. Soc. Jpn., 2006, 79, 177 CrossRef CAS; (c) S. Fukuzumi, Phys. Chem. Chem. Phys., 2008, 10, 2283 RSC; (d) S. Fukuzumi and K. Ohkubo, Coord. Chem. Rev., 2010, 254, 372 CrossRef CAS.
- (a) S. Fukuzumi and K. Ohkubo, J. Mater. Chem., 2012, 22, 4575 RSC; (b) S. Fukuzumi, K. Ohkubo, F. D'Souza and J. L. Sessler, Chem. Commun., 2012, 48, 9801 RSC.
- (a) K. Ohkubo and S. Fukuzumi, J. Porphyrins Phthalocyanines, 2008, 12, 993 CrossRef CAS; (b) K. Ohkubo and S. Fukuzumi, Bull. Chem. Soc. Jpn., 2009, 82, 303 CrossRef CAS.
- (a) S. Fukuzumi and T. Kojima, J. Mater. Chem., 2008, 18, 1427 RSC; (b) F. D'Souza and O. Ito, Chem. Commun., 2009, 4913 RSC; (c) F. D'Souza and O. Ito, Coord. Chem. Rev., 2005, 249, 1410 CrossRef CAS.
- (a) S. Fukuzumi, K. Saito, K. Ohkubo, T. Khoury, Y. Kashiwagi, M. A. Absalom, S. Gadde, F. D'Souza, Y. Araki, O. Ito and M. J. Crossley, Chem. Commun., 2011, 47, 7980 RSC; (b) S. Fukuzumi, K. Ohkubo, K. Saito, Y. Kashiwagi and M. J. Crossley, J. Porphyrins Phthalocyanines, 2011, 15, 1292 CrossRef CAS.
- S. Fukuzumi, I. Amasaki, K. Ohkubo, C. P. Gros, J.-M. Barbe and R. Guilard, RSC Adv., 2012, 2, 3741 RSC.
- (a) T. Kojima, T. Honda, K. Ohkubo, M. Shiro, T. Kusukawa, T. Fukuda, N. Kobayashi and S. Fukuzumi, Angew. Chem., Int. Ed., 2008, 47, 6712 CrossRef CAS; (b) S. Fukuzumi, T. Honda, K. Ohkubo and T. Kojima, Dalton Trans., 2009, 3880 RSC; (c) T. Kojima, K. Hanabusa, K. Ohkubo, M. Shiro and S. Fukuzumi, Chem.–Eur. J., 2010, 16, 3646 CrossRef CAS; (d) T. Honda, T. Nakanishi, K. Ohkubo, T. Kojima and S. Fukuzumi, J. Am. Chem. Soc., 2010, 132, 10155 CrossRef CAS.
- (a) F. D'Souza, E. Maligaspe, K. Ohkubo, M. E. Zandler, N. K. Subbaiyan and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 8787 CrossRef CAS; (b) F. D'Souza, N. K. Subbaiyan, Y. Xie, J. P. Hill, K. Ariga, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 16138 CrossRef CAS; (c) F. D'Souza, A. N. Amin, M. E. El-Khouly, N. K. Subbaiyan, M. E. Zandler and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 654 CrossRef CAS; (d) F. D'Souza and O. Ito, Chem. Soc. Rev., 2012, 41, 86 RSC.
- B. Grimm, J. Schornbaum, C. M. Cardona, J. D. van Paauwe, P. D. W. Boyd and D. M. Guldi, Chem. Sci., 2011, 2, 1530 RSC.
- (a) S. Fukuzumi, Pure Appl. Chem., 2007, 79, 981 CrossRef CAS; (b) S. Fukuzumi, in Functional Organic Materials, ed. T. J. J. Müller and U. H. F. Bunz, Wiley-VCH, 2007, pp. 465–510 Search PubMed.
- (a) L. Echegoyen, F. Diederich and L. E. Echegoyen, in Fullerenes: Chemistry, Physics and Technology, ed. K. M. Kadish and R. S. Ruoff, Wiley-Interscience, New York, 2000, pp. 1–51 Search PubMed; (b) D. M. Guldi and S. Fukuzumi, in Fullerenes: From Synthesis to Optoelectronic Properties, ed. D. M. Guldi and N. Martin, Kluwer, Dordorecht, 2003, pp. 237–265 Search PubMed.
- (a) S. Fukuzumi and D. M. Guldi, in Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, vol. 2, pp. 270–337 Search PubMed; (b) H. Imahori and S. Fukuzumi, Adv. Funct. Mater., 2004, 14, 525 CrossRef CAS.
- Y. Kashiwagi, K. Ohkubo, J. A. McDonald, I. M. Blake, M. J. Crossley, Y. Araki, O. Ito, H. Imahori and S. Fukuzumi, Org. Lett., 2003, 5, 2719 CrossRef.
- K. Ohkubo, H. Imahori, J. Shao, Z. Ou, K. M. Kadish, Y. Chen, R. K. Pandey, M. Fujitsuka, O. Ito and S. Fukuzumi, J. Phys. Chem. A, 2002, 106, 10991 CrossRef CAS.
- (a) K. Ohkubo, H. Kotani, J. Shao, Z. Ou, K. M. Kadish, G. Li, R. K. Pandey, M. Fujitsuka, O. Ito, H. Imahori and S. Fukuzumi, Angew. Chem., Int. Ed., 2004, 43, 853 CrossRef CAS; (b) S. Fukuzumi, K. Ohkubo, H. Imahori, J. Shao, Z. Ou, G. Zheng, Y. Chen, R. K. Pandey, M. Fujitsuka, O. Ito and K. M. Kadish, J. Am. Chem. Soc., 2001, 123, 10676 CrossRef CAS.
- L. Matrín-Gomis, K. Ohkubo, F. Fernández-Lázaro, S. Fukuzumi and Á. Sastre-Santos, J. Phys. Chem. C, 2008, 112, 17694 CrossRef CAS.
- F. D'Souza, R. Chita, K. Ohkubo, M. Tasior, N. K. Subbaiyan, M. E. Zandler, M. Rogacki, D. T. Gryko and S. Fukuzumi, J. Am. Chem. Soc., 2008, 130, 14263 CrossRef CAS.
- S. Fukuzumi, K. Ohkubo, H. Imahori and D. M. Guldi, Chem.–Eur. J., 2003, 9, 1585 CrossRef CAS.
- S. Fukuzumi, I. Nakanishi, T. Suenobu and K. M. Kadish, J. Am. Chem. Soc., 1999, 121, 3468 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834 CrossRef CAS.
- A. Takai, M. Chkounda, A. Eggenspiller, C. P. Gros, M. Lachkar, J.-M. Barbe and S. Fukuzumi, J. Am. Chem. Soc., 2010, 132, 4477 CrossRef CAS.
- (a) P. D. W. Boyd and C. A. Reed, Acc. Chem. Res., 2005, 38, 235 CrossRef CAS; (b) K. Tashiro and T. Aida, Chem. Soc. Rev., 2007, 36, 189 RSC; (c) D. Canevet, E. M. Pérez and N. Martín, Angew. Chem., Int. Ed., 2011, 50, 9248 CrossRef CAS.
- J.-M. Lehn, Supramolecular Chemistry, Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed.
- C. J. Chang, J. D. K. Brown, M. C. Y. Chang, E. A. Baker and D. G. Nocera, in Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, vol. 3, pp. 409–461 Search PubMed.
- (a) T. Hayashi and H. Ogoshi, Chem. Soc. Rev., 1997, 26, 355 RSC; (b) M.-J. Blanco, M. C. Jiménez, J.-C. Chambron, V. Heitz, M. Linke and J.-P. Sauvage, Chem. Soc. Rev., 1999, 28, 293 RSC; (c) I. Willner, E. Kaganer, E. Joselevich, H. Dürr, E. David, M. J. Günter and M. R. Johnston, Coord. Chem. Rev., 1998, 171, 261 CrossRef CAS.
- (a) H. Nobukuni, Y. Shimazaki, F. Tani and Y. Naruta, Angew. Chem., Int. Ed., 2007, 46, 8975 CrossRef CAS; (b) H. Nobukuni, F. Tani, Y. Shimazaki, Y. Naruta, K. Ohkubo, T. Nakanishi, T. Kojima, S. Fukuzumi and S. Seki, J. Phys. Chem. C, 2009, 113, 19694 CrossRef CAS; (c) H. Nobukuni, Y. Shimazaki, H. Uno, Y. Naruta, K. Ohkubo, T. Kojima, S. Fukuzumi, S. Seki, H. Sakai, T. Hasobe and F. Tani, Chem.–Eur. J., 2010, 16, 11611 CrossRef CAS.
- (a) H. Nobukuni, T. Kamimura, H. Uno, Y. Shimazaki, Y. Naruta and F. Tani, Bull. Chem. Soc. Jpn., 2012, 85, 862 Search PubMed; (b) H. Nobukuni, T. Kamimura, H. Uno, Y. Shimazaki, Y. Naruta and F. Tani, Bull. Chem. Soc. Jpn., 2011, 84, 1321 CrossRef CAS.
- (a) Y. Zeng, L. Biczok and H. Linchitz, J. Phys. Chem., 1992, 96, 5237 CrossRef CAS; (b) D. M. Guldi and M. Maggini, Gazz. Chim. Ital., 1997, 127, 779 CAS; (c) D. M. Guldi, H. Hungerbuehler, I. Carmichael, K.-D. Asmus and M. Maggini, J. Phys. Chem. A, 2000, 104, 8601 CrossRef CAS.
- (a) A. S. Aoyagi, E. Nishibori, H. Sawa, K. Sugimoto, M. Takata, Y. Miyata, R. Kitaura, H. Shinohara, H. Okada, T. Sakai, Y. Ono, K. Kawachi, K. Yokoo, S. Ono, K. Omote, Y. Kasama, S. Ishikawa, T. Komuro and H. Tobita, Nat. Chem., 2010, 2, 678 CrossRef CAS; (b) S. Aoyagi, Y. Sado, E. Nishibori, H. Sawa, H. Okada, H. Tobita, Y. Kasama, R. Kitaura and H. Shinohara, Angew. Chem., Int. Ed., 2012, 51, 3377 CrossRef CAS; (c) H. Okada, T. Komuro, T. Sakai, Y. Matsuo, Y. Ono, K. Omote, K. Yokoo, K. Kawachi, Y. Kasama, S. Ono, R. Hatakeyama, T. Kaneko and H. Tobita, RSC Adv., 2012, 2, 10624 RSC.
- S. Fukuzumi, K. Ohkubo, Y. Kawashima, D. S. Kim, J. S. Park, A. Jana, V. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2011, 133, 15938 CrossRef CAS.
- Y. Kawashima, K. Ohkubo and S. Fukuzumi, J. Phys. Chem. A, 2012, 116, 8942 Search PubMed.
- K. Ohkubo, Y. Kawashima and S. Fukuzumi, Chem. Commun., 2012, 48, 4314 RSC.
- Y. Matsuo, H. Okada, M. Maruyama, H. Sato, H. Tobita, Y. Ono, K. Omote, K. Kawachi and Y. Kasama, Org. Lett., 2012, 14, 3784 Search PubMed.
- Quasi-reversible CVs were observed in the electron oxidation processes because the adsorption on the electrode surface may occur due to the low solubility of the electrochemically oxidized porphyrin dimer.
- L. X. Chen, X. Zhang, E. C. Wasinger, K. Attenkofer, G. Jennings, A. Z. Murean and J. S. Lindsey, J. Am. Chem. Soc., 2007, 129, 9616 CrossRef CAS.
- The formation dynamics of the CS state [H4–CPDPy(OC6)˙++ Li+@C60˙−]) could not be observed by the femto- and nanosecond laser flash photolysis because the process would occur in the 10 ns–100 ns time range, which is out of the range of the present laser flash systems.
- In the case of the oligomeric porphyrin-fullerene systems, longer CS lifetimes (τcs = 0.70 and 0.84 ms) were observed due to the charge migration in the oligomeric arrays of the chromophores. See: S. Fukuzumi, K. Saito, K. Ohkubo, V. Troiani, H. Qiu, S. Gadde, F. D'Souza and N. Solladie, Phys. Chem. Chem. Phys., 2011, 13, 17019 Search PubMed.
- (a) R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265 Search PubMed; (b) R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155 CrossRef CAS.
- The decay of 3[Ni2–CPDPy(OC6)]* is much faster than electron transfer from 3[Ni2–CPDPy(OC6)]* to Li+@C60. The rate of electron transfer from 3[Ni2–CPDPy(OC6)]* to Li+@C60 is predicted as 106–107 s−1 because the driving force of electron transfer (−ΔGET = 0.30 eV) is similar to the case of electron transfer from Ni2–CPDPy(OC6) to 3[Li+@C60]* (−ΔGET = 0.33 eV, kET = 5.7 × 107 s−1).
- (a) Y. Zhang, C. Zhao, J. Yang, M. Kapiamba, O. Haze, L. J. Rothberg and M. -K. Ng, J. Org. Chem., 2006, 71, 9475 CrossRef CAS; (b) W. Huang, M. Wang, C. Du, Y. Chen, R. Qin, L. Su, C. Zhang, Z. Liu, C. Li and Z. Bo, Chem.–Eur. J., 2011, 17, 440 Search PubMed.
- C. Ruzié, L. Michaudet and B. Boitrel, Tetrahedron Lett., 2002, 43, 7423 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic and kinetic data. See DOI: 10.1039/c3sc22065f |
| This journal is © The Royal Society of Chemistry 2013 |