 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure elucidation and control of cyclic peptide-derived nanotube assemblies in solution†
Robert
Chapman
,
Ming Liang
Koh
,
Gregory G.
Warr
,
Katrina A.
Jolliffe
* and
Sébastien
Perrier
*
Key Centre for Polymers & Colloids, School of Chemistry, Building F11, The University of Sydney, NSW 2006, Australia. E-mail: kate.jolliffe@sydney.edu.au; sebastien.perrier@sydney.edu.au
First published on 5th April 2013
Abstract
We describe the solution assembly of polymer–cyclic peptide conjugates into nanotubes, by direct in situ measurements. The conjugates were assembled by exploiting the β-sheet assembly of the alt(D,L) cyclic octapeptide core. The conjugated polymer permits solubilization of the peptide and the resulting nanotubes, thus allowing for the first time the direct study of the assembly mechanism of this system. The resulting materials present unique properties for a wide range of applications. We find that the polymer can act to both shield the peptide core from the solvent and to put strain on the peptide core through steric repulsions. By controlling both the solvent mixture and the length of the polymer, control over the length of the resulting nanotubes can be obtained. In addition, monitoring the assembly with temperature allows the strength of the assembly to be probed, adding evidence for a co-operative mechanism of assembly. Finally, selective deuteration of the polymer component in SANS analyses leads to the precise measurement of the nanotubes core dimensions, and by cross-linking the polymeric shell, the structure of the nanotubes in solution are confirmed by transmission electron microscopy (TEM). This study establishes the fundamentals needed for the design and control of these new materials.
Introduction
The supramolecular stacking of cyclic peptides (CPs) has emerged in recent years as a remarkably versatile approach to the formation of organic nanotubes. The range of cyclic peptides that assemble in this way includes cycles containing 6–10 alternating D- and L-α amino acid residues,1 cyclic β peptides,2,3 and cyclic peptides containing alternating α- and γ- or α- and ε-residues.4–6 All such CPs adopt a rigid flat disc conformation, in which the amide bonds protrude perpendicular to the plane of the ring, and this allows them to stack through β-sheet hydrogen bonding to form a tube. The size of the cycle governs the internal diameter of the nanotube, which can therefore be tuned with great precision down to sub-nm sizes, an impossible scale to achieve with most systems. The choice of amino acid residues can be used to control the pore chemistry to a certain extent, as well as the external functionality of the nanotube through the side chains, which project equatorially from the core.7–9The versatility of this approach to nanotube formation has opened up much promise for applications as diverse as transmembrane ion channels and antibacterial agents, biosensors, and templates for nanowires.7,8 However, the full realization of such a great potential has been limited to date by two main drawbacks. The assembled cyclic peptide nanotubes (CPNTs) have limited solubility in all but the most polar solvents, due to their tendency to further aggregate into bundles of tubes. In addition, the length of the CPNT is very difficult to control. By functionalizing the periphery of the CP with polymers, the groups of Biesalski and Börner have independently shown that better solubility can be achieved,10,11 and steric bulk has also been shown to restrict the growth of the nanotubes, an observation that we also confirmed more recently.12–15 Our approach, which relies on the conjugation of polymeric chains to an individual cyclic peptide, permits isolation of the conjugates pre-assembly and provides an opportunity to control their organization into NTs. Indeed, the limited solubility of CPNTs not only restricts their application, but also makes the self-assembly very difficult to study. While the assembly of N-alkylated CPs into dimers has been extensively studied as a model system for the CPNTs,16 the association constants measured for these dimers are much lower than would be expected for the assembly of CPs into nanotubes, and thus they may not represent a good model for the assembly of non-alkylated CPs. Due to the solubility they impart, the attachment of polymers to CPs pre-assembly offers an ideal way to more directly study the factors that influence the self-assembly of the CPNTs in solution, in a way that has not previously been possible.
In this work, we study in situ the assembly of CPs modified with polymeric chains and investigate the factors that control their self-assembly into nanotubes with the support of small angle neutron scattering (SANS) and dynamic light scattering (DLS) techniques. We investigate the effect of polymer DP, solvent conditions, and temperature on the assembly, and for the first time extrapolate an enthalpy of association for the non-alkylated CPs. By deuterating parts of the conjugate we establish the structure of the nanotubes in solution, and by cross-linking the polymer shell to lock-in this structure, as we have shown recently on similar systems,17 we image the nanotubes in the dry state by TEM.
Materials and methods
Polymer synthesis
(Propynyl propanoate)yl butyltrithiocarbonate (PPBTC) was synthesized according to previous work in our group,14,18,19 and was mixed with deinhibited monomer, AIBN (0.1 mol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol PPBTC), and dioxane ([M] = 2.8 M). The mixture was degassed by bubbling with N2 for 15 min and heated in an oil bath at 70 °C, under N2 at 1 atm. Samples of the crude were taken throughout the reaction for determination of conversion by 1H-NMR, and the remainder was purified by precipitation and subsequent washing from an ice cold mixture of 10% water in methanol. Butyl acrylate was polymerized to DPs of 16, 30, 70, 90, 108, and 195 (as determined by 1H-NMR). Deuterated butyl acrylate (d9-BA) was polymerized to a DP of 29, and then chain extended with either 45 or 65 units of n-BA. Finally a BA/hydroxyethyl acrylate (HEA) random copolymer was prepared by mixing BA and HEA (9:1) with a total monomer:PPBTC:AIBN ratio of 40:1:0.1, and polymerizing as above. The product was determined by 1H-NMR to contain 31 BA units per chain, and 2.6 HEA units. All polymerizations were conducted to ∼80% conversion, and low dispersities were observed by SEC in all cases (see Table S1 and Fig. S1 in ESI†).
:1 mol PPBTC), and dioxane ([M] = 2.8 M). The mixture was degassed by bubbling with N2 for 15 min and heated in an oil bath at 70 °C, under N2 at 1 atm. Samples of the crude were taken throughout the reaction for determination of conversion by 1H-NMR, and the remainder was purified by precipitation and subsequent washing from an ice cold mixture of 10% water in methanol. Butyl acrylate was polymerized to DPs of 16, 30, 70, 90, 108, and 195 (as determined by 1H-NMR). Deuterated butyl acrylate (d9-BA) was polymerized to a DP of 29, and then chain extended with either 45 or 65 units of n-BA. Finally a BA/hydroxyethyl acrylate (HEA) random copolymer was prepared by mixing BA and HEA (9:1) with a total monomer:PPBTC:AIBN ratio of 40:1:0.1, and polymerizing as above. The product was determined by 1H-NMR to contain 31 BA units per chain, and 2.6 HEA units. All polymerizations were conducted to ∼80% conversion, and low dispersities were observed by SEC in all cases (see Table S1 and Fig. S1 in ESI†).
Conjugation
Cyclo[-D-Leu-L-Lys(N3)-D-Leu-L-Trp-]2, and cyclo[-D-Leu-L-Lys(N3)-]4, both 8 residue self-assembling cyclic peptides, were synthesized as previously in our group from azido lysine via solid phase peptide synthesis (SPPS).14 The peptide (15 mg, 0.013 mmol) was suspended in TFE (2.0 g) by sonication for 10 min, and to this was added a solution of the polymer (2.1 equiv.) and CuSO4 (4 equiv., 0.053 mmol, 13 mg) in DMF (2.0 g). Sodium ascorbate (0.053 mmol, 10 mg) was added, and the mixture was stirred in a microwave reactor at 100 °C for 15 min. The initial power input was set to 200 W and the vessel was cooled using N2 throughout the reaction to maximize the microwave power input, which averaged ∼50 W over the course of the reaction. The product was purified by dissolving the crude reaction mixture in DCM and washing it with EDTA (0.065 M, ×1), water (×2), and then brine. The organic phase was dried over MgSO4, and concentrated to dryness to yield the product. (pBA16)2–CP: 1H-NMR (300 MHz, d-TFA) δ ppm: 8.48 (s, 2H, triazole CH), 7.60 (s, 2H, Trp), 7.34 (s, 2H, Trp), 7.26–7.02 (m, 6H, Trp), 5.46 (s, 4H, O–CH2–triazole), 5.18 (s, 2H, Trp α H), 4.87–4.52 (m, 6H, Leu & Lys α H), 4.24 (s, 64H, O–CH2), 3.95 (s, 2H, CH2–S), 1.71 (s, 64H, O–CH2–CH2–) 1.42 (s, 64H, O–(CH2)2–CH2–), 0.98 (s, 96H, O–(CH2)3–CH3), 0.89–0.73 (m, 24H, Leu CH3), 0.45 (d, J = 10.15 Hz, 3H, RAFT C–CH3), 0.22–0.15 (m, 3H, S–(CH2)3–CH3). IR (ATR, ZnSe) cm−1: 3277, 2950–2850, 1733, 1687, 1624, 1541 (br).Cross-linking with toluene diisocyanate
The p(BA-co-HEA)–CP conjugate (0.05 g) was dissolved in DMF (0.5 ml) and diluted with distilled anhydrous THF to a final DMF concentration of 2% (v/v). 0.25 equivalents of toluene diisocyanate (TDI, 28 μmol) relative to the HEA sites was added dropwise from a stock solution of TDI (210 μmol, 30 μl) and dibutyltin dilaurate (6 μmol, 0.004 g) in anhydrous THF (5 ml). The mixture was stirred for 1 h or for 16 h under a nitrogen atmosphere over molecular sieves (4 Å) before quenching the reaction with 100 μl of water. The efficiency of the cross-linking was confirmed by concentrating a sample of the conjugate to dryness, redispersing it in DMF–TFA (3:1) and heating it for 1 h at 70 °C.
Self-assembly
Polymer–CP conjugates were assembled from 5 mM stock solutions in d-TFA or d-TFA–d7-DMF (1:9), which were heated for 2 h at 60 °C to ensure complete disaggregation. Nanotubes were assembled by dropwise addition of d8-THF to these stock solutions under vortex mixing. Samples in pure d7-DMF were heated overnight at 60 °C to ensure good dissolution of the conjugates. For the SANS studies, assembled conjugates were allowed to equilibrate for at least 72 h prior to measurement to ensure thermodynamic equilibrium.
Small angle neutron scattering (SANS)
With the exception of two samples, SANS was carried out on the Sans2d small-angle diffractometer at the ISIS Pulsed Neutron Source (STFC Rutherford Appleton Laboratory, Didcot, UK).20,21 A simultaneous q-range of 0.0045–0.75 Å−1 was achieved utilizing an incident wavelength range of 1.75–16.5 Å and employing an instrument set up of L1 = L2 = 4 m, with the 1 m2 detector offset vertically 150 mm and laterally 180 mm. The wave vector, q, is defined as:where θ is the scattered angle and λ is the incident neutron wavelength. Samples were prepared at a concentration of 0.5 mM in deuterated solvents, providing the necessary contrast and were contained in 2 mm path length Hellma quartz cells. Each raw scattering dataset was corrected for the detector efficiencies, sample transmission and background scattering and converted to scattering cross-section data (Σ/∂Ω vs. q) using the instrument-specific software.22 SANS of the two conjugates in TFA–DMF–THF (1
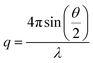 :9:90) were performed on the 18 m SANS instrument at the HANARO reactor at the Korean Atomic Energy Research Institute (KAERI). Scattering was taken from 2 mm path length cells with 5.14 Å neutrons onto a 128 × 128 pixel, 640 × 640 mm2 detector at 1.3 m and 9.0 m sample to detector distances, giving a combined q range of 0.008–0.3 Å−1.
:9:90) were performed on the 18 m SANS instrument at the HANARO reactor at the Korean Atomic Energy Research Institute (KAERI). Scattering was taken from 2 mm path length cells with 5.14 Å neutrons onto a 128 × 128 pixel, 640 × 640 mm2 detector at 1.3 m and 9.0 m sample to detector distances, giving a combined q range of 0.008–0.3 Å−1.
Each raw scattering dataset was corrected for the detector efficiencies, sample transmission and background scattering and converted to scattering cross-section data (∂Σ/∂Ω vs. q) using the instrument-specific software.22 These data were placed on an absolute scale (I(q)) using the scattering from a standard sample (a solid blend of hydrogenous and predeuterated polystyrene) in accordance with established procedures.23
Dynamic light scattering (DLS)
DLS experiments were performed with a 633 nm wavelength laser at 90° with a 400 μm spot size in a decalin bath at 25 °C. Correlation functions were fit using Brookhaven software with a CONTIN model. All solvents were filtered through a 20 nm glass microfibre filter (Whatman Anotop) prior to use to eliminate dust.Transmission electron microscopy (TEM)
TEM images were taken on a JEOL JEM-1400 microscope at 120 kV. Sample grids were prepared by coating 400 mesh copper grids with a Formvar film, and sputter coating with carbon (∼10 nm). Samples were deposited on the grids from solution, and dried prior to imaging.Results and discussion
In this work we have investigated the mechanisms by which the nanotubes assemble and how the molecular weight of the polymer affects the nanotube formation. A cyclic peptide based on our previous work was synthesized containing two azide sites.14 A range of butyl acrylate chains with degrees of polymerization (DPs) ranging from 16 to 195 were conjugated to this peptide to generate a family of conjugates ((pBA16)2–CP to (pBA195)2–CP, Fig. 1). In all cases 5% excess polymer was used in the synthesis ensuring 100% conversion. This was confirmed by 1H-NMR, the complete disappearance of the azide signal by IR and a clear shift in molecular weight distribution by SEC (see Fig. S1 and S2 in ESI†). By taking this convergent synthesis approach to the preparation of the polymer–CP conjugate, any interference of unconjugated peptide on the assembly was eliminated.18,19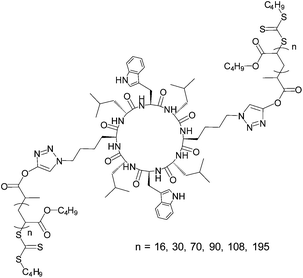 | ||
| Fig. 1 Structure of assembling (pBAn)2–CP conjugates. | ||
The ability of the cyclic peptide to form strong β-sheet networks, even when conjugated to the polymer, is critical if the peptide is to guide the formation of the nanotube, and has been established by us and others in previous work. The conformation of the peptide can be most easily determined by studying the position of the vibrational stretching mode from the amide group in the fingerprint region of an infra-red (IR) spectrum.24–26 In both the unconjugated cyclic peptide, and across all the conjugates presented herein, characteristic anti-parallel β-sheet amide stretching signals were observed in the dried state on an ATR crystal (Fig. 2b) at 1624 and 1687 cm−1 corresponding well to the β-sheet amide I regions, and at 1541 cm−1 corresponding to the expected amide II band. In the polymer conjugates, an additional broad signal can be seen around 1650 cm−1 consistent with the random coil structure expected from the polymer.
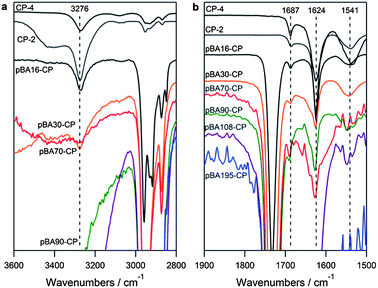 | ||
| Fig. 2 ATR-IR of cyclic peptides and (pBAn)2–CP conjugates after precipitation from water (a) in the amide A N–H stretching region and (b) in the amide I and II stretching region. | ||
IR can also be used to estimate the inter-ring stacking distance, by a Krimm's analysis of the N–H stretch in the amide A band between 3225 and 3280 cm−1.27 All alt(L,D) 8 α residue cyclic peptides synthesized in the literature have shown a stretch of 3277 cm−1 in the assembled state, corresponding to an N–H⋯O distance of 2.85 Å and an inter-subunit spacing of 4.71 Å, in good agreement with crystal structure data for these materials.9 As can be seen in Fig. 2a, our cyclic peptide exhibits an N–H stretch at almost exactly the same position, 3276 cm−1, indicating the same inter-ring stacking distance and conjugation of pBA of DP 16, 30, or 70 does not cause any shift in this peak. While analysis of higher DP polymers is difficult because the peptide signals become very small compared to the polymer and the traces become very noisy, this is a good indication that the polymer does not significantly affect the inter-ring spacing of the peptide.
Controlling the dimensions of the nanotubes in solution
Confident of the strength of the peptide interactions from the IR studies, we proceeded to investigate the self-assembly properties of (pBAn)2–CP conjugates in solution. The formation of nanotubes was characterized by a combination of dynamic light scattering (DLS), small angle neutron scattering (SANS) and transmission and scanning electron microscopy (TEM and SEM) on the dried films. The hydrogen bonding of these conjugates is remarkable strong, even with the polymer attached. In order to study the self-assembly process we first had to establish conditions under which the conjugates were not aggregated. This was achieved by dissolving the conjugates in neat TFA and heating to 70 °C. The dissociation of the tubes can be monitored by the falling count rate with time in dynamic light scattering. Even in very competitive solvents such as neat TFA, it takes several hours to completely disaggregate the tubes. The ester linkage on the RAFT agent by which the polymer is attached to the peptide in the conjugate is liable to acid cleavage, so it is important to note that the drop in count rate is due to the dissociation of the peptide and not the disintegration of the conjugate. We saw no evidence of degradation of either the RAFT agent or a DP16 pBA by 1H-NMR after 2 h at 60 °C. To ensure the ester group is still stable when close to the triazole group after conjugation, we analysed a model system in which benzyl azide was reacted with the RAFT agent, and again saw no hydrolysis of the ester group over 2 h at 60 °C (see Fig. S3–S5 in ESI†).As samples were diluted from their neat TFA solution with DMF, THF or CH2Cl2, all of which compete less effectively than TFA for the hydrogen bonding sites on the peptide, the DLS count rate increased and the formation of aggregates was observed (see Fig. S6 and S7 in ESI†). Diluting with THF or CH2Cl2 had a more pronounced effect than did dilution with DMF, yielding a distribution of large aggregates around 200–500 nm in hydrodynamic diameter at ratios of THF or CH2Cl2:TFA of 1:9 (v/v) and higher. Higher THF content resulted in longer assemblies, as evidenced by a much higher count rate. Evidence of the structure of these aggregates can be seen by TEM and SEM of the dried films, as we have shown previously.14 However, because these techniques are conducted in the dried state it is impossible to be sure that what is imaged is actually representative of the structures present in solution, and have not changed during the drying process. We therefore turned to small angle neutron scattering (SANS) to characterize the nanotubes in solution. SANS has been used to great effect in the characterisation of similar peptide–polymer nanostructures.28–30 Rod-like architectures, such as those expected in our case are known to display a q−1 intensity dependence over the range of characteristic lengths radius < q−1 < length.31,32 Because longer tubes display this power law behaviour to lower q than shorter tubes, comparison of different length tubes from the scattering data is relatively accurate, making SANS an ideal technique for the investigation of the self-assembly of this system. When the (pBA90)2–CP conjugates were dissolved in a mixture of DMF and TFA (3:1 v/v) in which assembly was not expected, very small aggregates were observed (Fig. 3a). The shape of the scattering pattern is quite different from what would be expected from a spherical or rod-like aggregate, but fits very well to a stacked disc model in which three discs roughly 7 nm in radius are stacked together (see Fig. S8 in ESI†). Such dimensions are commensurate with a trimer of stacked conjugate rings. At the same concentration of the (pBA90)2–CP conjugate in 100% DMF, or in TFA–THF (1:9, Fig. 3a), the resulting scattering pattern is characteristic of long cylinders, as evidenced by the q−1 dependence of the scattered intensity at low scattering angles. These data were fitted to a core–shell–shell cylinder model adapted from a core–shell cylinder form factor, using the NIST macros in IGOR Pro (see Fig. S8–S13 in ESI†).33 Interactions were neglected at these dilutions, and any effect from the small amount of free (unconjugated) polymer was ignored as this was expected to be negligible. We fit to a double-shell model in order to allow for a more gradual change in scattering length density (SLD) arising from solvent penetration into the polymer shell of the nanotube in solution, and therefore more accurately model the ‘soft’ interface expected from highly solvated tubes. This model yielded excellent fits to the scattering data obtained from all of the conjugates at a concentration of 0.5 mM in DMF or in TFA–THF (1:9), confirming the presence of tubes in these solvents. The outer shells of the tubes were indeed found to be highly solvated, as seen by the relatively small difference between their SLD and that of the solvent. The fitted volume fractions of the conjugates is higher than that of the dry polymer in solution due to this high solvation. Diluting the conjugates from 0.5 mM to 0.1 mM in either solvent system reduced the scattering intensity but did not affect the dimensions of the tubes, indicating that 0.5 mM is well above the critical aggregation concentration even in a competitive solvent such as DMF.
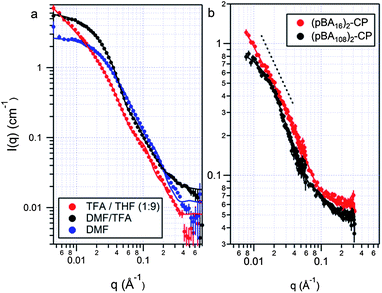 | ||
| Fig. 3 SANS data and fits for (a) pBA90–CP conjugates assembled in DMF, TFA–THF (1:9) and TFA–DMF (1:3); and (b) pBA16–CP and pBA108–CP conjugates assembled in TFA–DMF–THF (1:9:99). A core–shell–shell cylinder model was used in all cases except for the conjugates in TFA–DMF (1:3), where a stacked disc model was used. | ||
We were interested in using SANS to investigate the effect of the polymer DP on the assembly of the conjugate in solution. We hypothesized that increasing the DP of the polymer should shorten the nanotubes due to the increased steric bulk, in line with the work of Biesalski and Couet,13 and our previous results with this system.14 To minimize competition from the solvent, we prepared assemblies by dissolving the conjugates in a TFA–DMF (1:9) mixture, and diluting with THF to a final TFA concentration of 1%. Fig. 3b compares SANS profiles for a long and short polymer conjugate assembled in this fashion. The (pBA16)2–CP conjugate formed tubes, >150 nm in length, and 10 nm in diameter. These nanotubes are too long to be more accurately measured as the scattering is still increasing as q−1 at the lowest accessible angle (q). Assuming that the hydrogen bonding distance of this conjugate is similar to that of the unconjugated peptide, a fair assumption based on the IR discussed above, such a length represents an aggregation number >300. As expected, the (pBA108)2–CP conjugates formed much shorter tubes approximately 36 nm in length, corresponding to an aggregation number of ∼75 (Fig. 4). This confirms that the dimensions of the nanotube in such weakly competitive solvents are governed by the steric bulk of the polymer. Because of the polymer bulk, fewer (pBA108)2–CP than (pBA16)2–CP conjugates are able to stack before any subsequent elongation is blocked. The diameter of the (pBA108)2–CP nanotubes was found to be just under double that of the (pBA16)2–CP nanotubes, commensurate with what would be expected from the longer polymer.
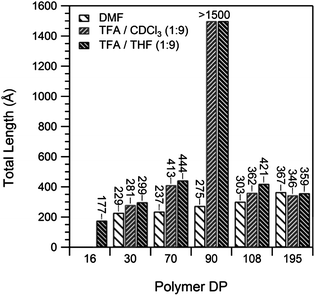 | ||
| Fig. 4 Length of pBA–CP nanotubes from the SANS data, fit to a core–shell–shell cylinder model in various solvent mixtures. | ||
In a more strongly competing solvent the DP of the polymer has a remarkably different effect on the length of the nanotubes. Fig. 4 summarizes the nanotube lengths obtained from the SANS data from conjugates with a polymer of DP 16–195 assembled in 100% DMF. Unlike with the sample with 1% TFA above, increasing polymer DPs resulted in longer nanotube assemblies. The length of the tubes almost doubled, from 23 nm for the (pBA30)2–CP conjugates up to 37 nm for the (pBA195)2–CP conjugates, with radii of around 10 nm in all cases, indicating that a second effect apart from steric hindrance governs the tube dimensions in such systems. We attribute this increase in length to increased shielding of the hydrogen bonding peptide centre from the competitive DMF solvent by the polymer as the length of the chain is increased. Short polymers should be relatively inflexible, and unable to protect the core from the DMF, but longer polymers are able to provide a better shell around the assembled core.
When the conjugates were assembled in a solvent mixture of intermediate hydrogen bonding competitiveness, a combination of both effects was observed in which both the shielding of the core by the polymer, and the steric bulk of the polymer affected nanotube assembly. The SANS patterns of the pBA conjugates in 1:9 TFA–THF (Fig. 4) show that nanotube lengths increase with increasing polymer DP up to the DP 90 conjugate, and then decrease as the polymer DP was further increased to 108 and 195. This suggests that up to a DP of 90, shielding of the peptide core from the competitive solvent governs the length of the nanotubes. Larger polymers, as was the case in 100% DMF, are more effective at shielding the core. Above this DP, however, the extra bulk of the polymer opposes assembly, preventing long assemblies from forming. It is important to note that all samples were measured at thermodynamic equilibrium and were not under kinetic control. This was determined by mixing separate samples of tubes from the DP 90 conjugates with tubes from a deuterated DP 29 conjugate in TFA–THF (1:9). No difference between SANS spectra measured at 10 min and after 4 days after mixing was observed, indicating any kinetic effects are very short lived.
Very similar results were seen in d-chloroform (Fig. 4). Despite its much lower scattering length density (3.16 × 10−6versus 6.35 × 10−6 Å−2 for d8-THF), leading to much weaker contrast against the hydrogenous (pBA16)2–CP and consequently lower quality data, assemblies of the pBA–CP conjugates of DP 30–195 could be fitted and show the same trend in tube length vs. polymer DP as in TFA–THF (1:9, Fig. 4). In both solvents DP 90 polymers provide the right balance between core shielding and chain volume to allow long assemblies. Small polymers failed to provide enough shielding, and longer polymers were too bulky.
Probing the strength of assembly
In order to probe the strength of the assemblies in solution further, we investigated the effect of temperature by heating the conjugates in 100% DMF from 25 to 70 °C. Because the tubes were very short in DMF at 25 °C, we anticipated that any temperature effect on the assembly should be most visible in this case. Each sample was held at 70 °C for 20 min prior to measuring the DLS, and for 10 h prior to measuring the SANS profile to ensure complete equilibration. The volume fraction and the shell SLD's were held constant in the SANS modeling of each conjugate between each temperature to allow a direct comparison of the lengths. Interestingly, the DLS results (see Fig. S7 in ESI†) show very little difference between 70 and 20 °C. The count rate even drops slightly as the samples were cooled, contrary to what might be expected if the aggregates were increasing in size at lower temperatures.SANS also revealed only a change of a few nanometers in the length of the tubes upon heating (Fig. 5) from 25 to 70 °C. Even the greatest observed change (pBA195–CP) of 6 nm corresponds to only ∼12 conjugate units, and though this length change is small, it lies well within the error from the model fitting (<1 nm in all cases, see ESI†). Such a small change in length confirms that the interaction is indeed remarkably strong, but is sufficient to allow an estimate the strength of the interaction. These nanotubes can be regarded as living polymers similar to worm-like micelles, which we can describe by co-opting the theoretical work of Israelachvili et al.34 The relationship between the average length or aggregation number of a cylindrical aggregate, the total concentration of assembled species, S, and the energy cost of the end cap, ΔG0end-cap, is given by:
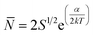
| α = (μ0end-cap − μ0cylinder)Nend = 2ΔG0end-cap |
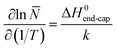
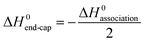
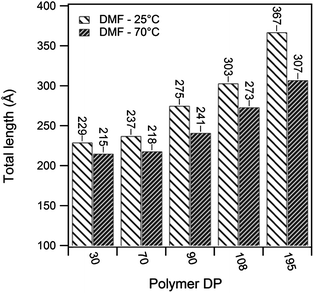 | ||
| Fig. 5 Total tube lengths for (pBAn)2–CP conjugates assembled in d7-DMF at 25 and 70 °C, based on the SANS data fit to a core–shell–shell cylinder model. | ||
As shown in Table 1 the enthalpy of association in DMF was found to be −2 to −6 kJ mol−1. Such values are an order of magnitude lower than the enthalpies determined for similar unconjugated N-methylated CPs in CDCl3,35 but are still surprisingly high given the conjugation of the polymer and the high hydrogen bonding propensity of DMF compared to CDCl3. The enthalpy of association was seen to increase with increasing polymer DP due to the shielding effect of the polymer in DMF as discussed earlier. The temperature and concentration of the assembling motif are the two variables that have typically been used to investigate the mechanism of supramolecular polymerizations.36 As shown above, however, these variables had only a limited effect on the assembly of this system within ranges that could be observed by SANS, which indicates the strength of the CP–CP interaction. Most anti-parallel β-sheet hydrogen bonding systems, including Ghadiri's peptide assemblies, have typically been assumed to assemble through either co-operative or anti-cooperative mechanisms, due to the high stability of the aggregates.6,7,24,37 The observed stability is inconsistent with the relatively low association constants (<3000 M−1) measured for the N-alkylated dimers,16 suggesting that the association constants for the initial nucleation period is not the same as that of the subsequent elongation. Recent experimental computational studies support the idea that this class of assembling cyclic peptides polymerize via a co-operative mechanism.38–40 Qu et al. used the semi-empirical molecular orbital method AM1 and the density functional methods B3LYP and PBE1PBE to propose that the L–L antiparallel dimer and not individual cyclic peptide units are the basic building block of CP nanotubes.38
| Conjugate | ΔH0end (kJ mol−1) | ΔH0association (kJ mol−1) |
|---|---|---|
| p(BA30)2–CP | 1.12 | −2.24 |
| p(BA70)2–CP | 1.59 | −3.18 |
| p(BA90)2–CP | 2.53 | −5.06 |
| p(BA108)2–CP | 1.93 | −3.86 |
| p(BA195)2–CP | 3.36 | −6.72 |
The SANS, DLS and TEM results from our polymer–peptide conjugate systems also support the conclusion that the assembly is co-operative and not isodesmic. The high ΔH0association, and the length of the assembly (>300 units in THF or CDCl3) confirms that the association constants, at least for elongation, are far stronger than those observed for the N-methylated dimers. In addition, the sharp transition between the unassembled and assembled states that was observed upon dilution of a TFA solution with DMF is consistent with an anti-cooperative growth mechanism.41 The presence of a small dimer or trimer in DMF–TFA (3:1) prior to assembly of the nanotube is perhaps indicative of a very small ‘seed’ nanotube prior to elongation.
Internal structure of the nanotubes by selective deuteration
In order to gain a better understanding of the structure of the nanotube cores, a deuterated DP 29 pBA–CP conjugate was prepared to match the SLDs of the polymer and (deuterated) solvent, rendering the polymer sheath virtually invisible by SANS (see Fig. S14 in ESI†). The SANS pattern for the [p(d9-BA)29]2–CP conjugate in n-THF–d-TFA (9:1) is compared to (pBA30)2–CP in d8-THF–d-TFA (9:1) in Fig. 6a. The fitted length of the tubes are identical, but deuteration of the polymer shell highlights the hydrogenous peptide core and reveals its radius to be about 5 Å, as expected from crystal structures of similar unconjugated peptides.
![SANS patterns and fits for (a) [p(d9-BA)29]2–CP tubes in n-THF–d-TFA (9 : 1) compared to (pBA30)2–CP tubes in d8-THF–d-TFA (9 : 1) and (b) for (pBA70)2–CP tubes with deuterated and hydrogenous cores in d8-THF–d-TFA (9 : 1).](/image/article/2013/SC/c3sc00064h/c3sc00064h-f6.gif) | ||
| Fig. 6 SANS patterns and fits for (a) [p(d9-BA)29]2–CP tubes in n-THF–d-TFA (9:1) compared to (pBA30)2–CP tubes in d8-THF–d-TFA (9:1) and (b) for (pBA70)2–CP tubes with deuterated and hydrogenous cores in d8-THF–d-TFA (9:1). | ||
The convergent approach to the preparation of CP–polymer nanotubes we have employed makes it relatively simple to prepare nanotubes from block copolymers, resulting in concentric tubes. In theory, the inner shell could be made from a different functionality to the outer shell, and the diameters can be tuned independently by altering the DP of each block. However, characterization of such a block copolymer nanotube is very difficult, as it is almost impossible to observe the inner block by any microscopy technique. However, by selectively deuterating one block, the internal structure of the nanotube can be observed by SANS. We have demonstrated proof of principle of this by chain extension of the p(d9-BA)29 macroRAFT agent with hydrogenous n-BA. Subsequent conjugation and assembly yielded two nanotube architectures with an inner deuterated DP 29 core and outer shells of DPs 45 and 65 to compare with the fully hydrogenous DP 70 and 90 conjugates, respectively. As can be seen in Fig. 6b, deuterating the inner block drastically alters the scattering pattern compared to the fully hydrogenous (pBA70)2–CP and (pBA90)2–CP. The model fits (see Fig. S15 and S16 in ESI†) reveal that the inner and outer shells of the nanotubes prepared by chain extension are indeed well segregated into deuterated inner and hydrogenous outer shells. While the structure of the two blocks here are chemically identical, there is no reason why this technique could not be used to prepare inner and outer shells with different functionalities.
Cross-linking the nanotubes to confirm the structure
By cross-linking the tubes we were able to use TEM to confirm the structure of the tubes observed in solution by SANS, without any risk of changes to the structure upon drying. Beyond confirmation of the structure of the nanotubes, the ability to stabilize polymeric nanotubes through cross-linking is useful from a materials perspective. A nice example of cross-linking these type of tubes has been shown by Biesalski and coworkers, who polymerized ethylene glycol dimethacrylate (EGDMA) divergently from the surface of a preassembled tube.42 In addition to providing stable nanotubes such as these, our approach makes it possible to tune the length of the tubes prior to locking in the structure with the cross-linking reaction. Because of the dynamic nature of the assembly, a very fast cross-linking reaction is required, such that the cross-linking of the tubes occurs faster than the exchange of the peptide–polymer conjugates between tubes. For this reason toluene diisocyanate was chosen as a cross-linker. Not only do isocyanates react extremely quickly with a range of nucleophiles,43 particularly in the presence of catalytic amounts (0.01 equiv.) of dibutyltin dilaurate,44,45 but they also reach quantitative conversion under mild conditions and would not be expected to cleave the thioester bond on the RAFT agent.46Isocyanate reactive OH functionality was introduced into the shell of the tubes by incorporating hydroxyethyl acrylate (HEA) into a polymer of BA. Incorporation of 10% HEA relative to total monomer resulted in highly uniform polymers containing 2.6 HEA units and 31 BA units. This polymer was attached to the peptide in the same way as the pure pBA polymers (see Fig. S17 in ESI†). The conjugate was assembled in DMF–THF (2:98), and cross-linked by dropwise addition of a solution of toluene diisocyanate (TDI) in THF. DLS revealed that the cross-linked tubes were stable in mixtures of DMF–TFA (3:1) which caused disaggregation of an uncross-linked sample (see Fig. S18 in ESI†). Imaging with TEM revealed the presence of the expected rod-like structures (Fig. 7), confirming the validity of the SANS model.
 | ||
| Fig. 7 Transmission electron micrographs of 0.5 mg ml−1 conjugate after cross-linking with 0.5 equiv. TDI. Quenched after 16 h and deposited on grid from THF. Grids unstained. | ||
Conclusions
We have used polymer conjugation and SANS to elucidate the in situ structure of nanotubes formed by the assembly of cyclic peptides in solution. Two factors were determined to control the length of the tubes. In solvent mixtures that strongly hydrogen bond to the cyclic peptide, the polymer acts to shield the core and increasing the DP of the polymeric chain results in increasing length of the nanotube. In solvent mixtures that are less competitive, the reverse is true and increasing the polymer DP reduces the length of the nanotubes through steric strain. By measuring the effect of temperature on the length of the nanotubes we were able to determine the enthalpy of assembly for the conjugates to be ∼1 to 3 kJ mol−1 in DMF, and by selective deuteration of the inner shell of the nanotubes, the size of the core could be studied. Finally we have shown the cross-linking of the shell using a diisocyanate to stabilize the nanotubes and confirm their structure by TEM. These results improve the mechanistic understanding of how this class of cyclic peptides assemble, and lays the path to new materials in which the polymer is used to not only functionalize, but also influence the assembly of peptides into nanotubes by enabling control over the dimensions of the assembled structure.Acknowledgements
The authors thank ISIS and KAERI for neutron scattering beamtime and AINSE for financial support to conduct the SANS experiments. The authors would like to thank Dr Sarah Rogers at ISIS for instrument training and support, and Dr Paul Fitzgerald for the samples run at HANARO. Algi Serelis from DuluxGroup is thanked for provision of the precursor RAFT agent. The Australian Research Council is acknowledged for funding (Discovery Program), and R. C. and M. L. K. thank the Australian government for the provision of an Australian Postgraduate Award.Notes and references
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS.
- D. Seebach, J. L. Matthews, A. Meden, T. Wessels, C. Baerlocher and L. B. McCusker, Helv. Chim. Acta, 1997, 80, 173–182 CrossRef CAS.
- T. D. Clark, L. K. Buehler and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120, 651–656 CrossRef CAS.
- M. Amorin, L. Castedo and J. R. Granja, J. Am. Chem. Soc., 2003, 125, 2844–2845 CrossRef CAS.
- J. H. van Maarseveen, W. S. Horne and M. R. Ghadiri, Org. Lett., 2005, 7, 4503–4506 CrossRef CAS.
- W. S. Horne, C. D. Stout and M. R. Ghadiri, J. Am. Chem. Soc., 2003, 125, 9372–9376 CrossRef CAS.
- D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988–1011 CrossRef CAS.
- R. J. Brea, C. Reiriz and J. R. Granja, Chem. Soc. Rev., 2010, 39, 1448–1456 RSC.
- R. Chapman, M. Danial, M. L. Koh, K. A. Jolliffe and S. Perrier, Chem. Soc. Rev., 2012, 41, 6023–6041 RSC.
- J. Couet, J. D. Jeyaprakash, S. Samuel, A. Kopyshev, S. Santer and M. Biesalski, Angew. Chem., Int. Ed., 2005, 44, 3297–3301 CrossRef CAS.
- M. G. J. ten Cate, N. Severin and H. G. Börner, Macromolecules, 2006, 39, 7831–7838 CrossRef CAS.
- J. Couet and M. Biesalski, Soft Matter, 2006, 2, 1005–1014 RSC.
- J. Couet and M. Biesalski, Small, 2008, 4, 1008–1016 CrossRef CAS.
- R. Chapman, K. A. Jolliffe and S. Perrier, Polym. Chem., 2011, 2, 1956–1963 RSC.
- R. Chapman, G. G. Warr, S. Perrier and K. A. Jolliffe, Chem.–Eur. J., 2013, 19, 1955–1961 CrossRef CAS.
- T. D. Clark, J. M. Buriak, K. Kobayashi, M. P. Isler, D. E. McRee and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120, 8949–8962 CrossRef CAS.
- R. Chapman, K. A. Jolliffe and S. Perrier, Adv. Mater., 2013, 25, 1170–1172 CrossRef CAS.
- R. Chapman, K. A. Jolliffe and S. Perrier, Aust. J. Chem., 2010, 63, 1169–1172 CrossRef CAS.
- C. K. Poon, R. Chapman, K. A. Jolliffe and S. Perrier, Polym. Chem., 2012, 3, 1820–1826 RSC.
- STFC Rutherford Appleton Laboratory, Didcot, UK, http://www.isis.stfc.ac.uk.
- R. K. Heenan, S. E. Rogers, D. Turner, A. E. Terry, J. Treadgold and S. M. King, J. Neutron Res., 2011, 22, 19–21 Search PubMed.
- Mantid Project, http://www.mantidproject.org.
- G. D. Wignall and F. S. Bates, J. Appl. Crystallogr., 1987, 20, 28 CrossRef CAS.
- J. D. Hartgerink, J. R. Granja, R. A. Milligan and M. R. Ghadiri, J. Am. Chem. Soc., 1996, 118, 43–50 CrossRef CAS.
- P. I. Haris and D. Chapman, Biopolymers, 1995, 37, 251–263 CrossRef CAS.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39, 549–559 CrossRef CAS.
- A. Lautié, F. Froment and A. Novak, Spectrosc. Lett., 1976, 9, 289–299 CrossRef.
- A. Lübbert, V. Castelletto, I. W. Hamley, H. Nuhn, M. Scholl, L. Bourdillon, C. Wandrey and H.-A. Klok, Langmuir, 2005, 21, 6582–6589 CrossRef.
- I. W. Hamley, M. J. Krysmann, A. Kelarakis, V. Castelletto, L. Noirez, R. A. Hule and D. J. Pochan, Chem.–Eur. J., 2008, 14, 11369–11375 CrossRef CAS.
- H. G. Börner, B. M. Smarsly, J. Hentschel, A. Rank, R. Schubert, Y. Geng, D. E. Discher, T. Hellweg and A. Brandt, Macromolecules, 2008, 41, 1430–1437 CrossRef.
- C. Moitzi, N. Freiberger and O. Glatter, J. Phys. Chem. B, 2005, 109, 16161–16168 CrossRef CAS.
- O. Glatter and O. Kratky, Small Angle X-ray Scattering, Academic, London, 1982 Search PubMed.
- S. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- M. R. Ghadiri, K. Kobayashi, J. R. Granja, R. K. Chadha and D. E. McRee, Angew. Chem., Int. Ed. Engl., 1995, 34, 93–95 CrossRef CAS.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem.–Eur. J., 2010, 16, 362–367 CrossRef CAS.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- W. Qu, H. Tan, G. Chen and R. Liu, Int. J. Quantum Chem., 2010, 110, 1648–1659 CAS.
- R. Vijayaraj, S. Sundar Raman, R. Mahesh Kumar and V. Subramanian, J. Phys. Chem. B, 2010, 114, 16574–16583 CrossRef CAS.
- R. Vijayaraj, S. Van Damme, P. Bultinck and V. Subramanian, J. Phys. Chem. B, 2012, 116, 9922–9933 CrossRef CAS.
- P. A. Korevaar, C. Schaefer, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482–13491 CrossRef CAS.
- R. Gokhale, J. Couet and M. Biesalski, Phys. Status Solidi A, 2010, 207, 878–883 CrossRef CAS.
- E. Dyer, H. A. Taylor, S. J. Mason and J. Samson, J. Am. Chem. Soc., 1949, 71, 4106–4109 CrossRef CAS.
- F. Biedermann, E. A. Appel, J. del Barrio, T. Gruendling, C. Barner-Kowollik and O. A. Scherman, Macromolecules, 2011, 44, 4828–4835 CrossRef CAS.
- G. Gody, C. Rossner, J. Moraes, P. Vana, T. Maschmeyer and S. Perrier, J. Am. Chem. Soc., 2012, 134, 12596–12603 CrossRef CAS.
- J. Moraes, T. Maschmeyer and S. Perrier, Aust. J. Chem., 2011, 64, 1047–1053 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation data for the conjugates, as well as detailed SANS data and model fitting parameters. See DOI: 10.1039/c3sc00064h |
| This journal is © The Royal Society of Chemistry 2013 |