 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Double-channel photosystems with antiparallel redox gradients: templated stack exchange with porphyrins and phthalocyanines†
Giuseppe
Sforazzini
,
Raluca
Turdean
,
Naomi
Sakai
and
Stefan
Matile
*
Department of Organic Chemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Web: www.unige.ch/sciences/chiorg/matile/ Fax: +41 22 379 3215; Tel: +41 22 379 6523
First published on 19th February 2013
Abstract
We report the synthesis of multicomponent surface architectures composed of phthalocyanines (Pc), porphyrins (TPP) and naphthalenediimides (NDI). Naphthalenediimide stacks are grown first by self-organizing surface initiated disulfide-exchange polymerization (SOSIP). An oriented redox gradient driving electrons toward the surface is applied by growing electron-richer NDI stacks on top of poorer ones. Lateral stacks of porphyrins and phthalocyanines are then added by templated stack exchange (TSE). A three-component gradient is constructed to drive the holes away from the solid surface. Antiparallel gradients are found to minimize charge recombination during photocurrent generation. Templates used for stack exchange also serve as hole barriers, whereas their size has surprisingly little importance. These results demonstrate the compatibility of SOSIP-TSE technology with porphyrins and phthalocyanines, confirm the importance of oriented antiparallel gradients to minimize charge recombination, and show that electronics rather than the size matter to template stack exchange.
Supramolecular architectures of the highest sophistication account for biological function. The question of what we would get if organic chemists could succeed in building functional systems of similar complexity with similar precision is attracting much scientific attention.1–3 To contribute new approaches to answer this fundamental question, we focused earlier on multifunctional photosystems in lipid bilayers4 but quickly moved on to learning how to grow multicomponent architectures directly on solid surfaces.5,6 This is important to preserve the directionality needed to build oriented gradients as in biological photosystems.
A zipper assembly, a sticky-end layer-by-layer approach, has been introduced first for this purpose.5 To achieve similar sophistication with less demanding synthetic efforts, self-organizing surface-initiated polymerization (SOSIP) was conceived next.6 SOSIP affords oriented π-stacks on oxide surfaces that are embedded in hydrogen-bonded networks and poly(disulfide) backbones.6,7 To build double-channel photosystems, templated self-sorting (TSS)8 and templated stack exchange (TSE) have been introduced as complementary methods.9 TSS is a supramolecular approach that aims to use self-sorting during co-SOSIP to transcribe information from mixed monolayers into 3D architectures. TSE, a more robust covalent approach, operates with non-interfering templates and hydrazones during SOSIP that can be replaced afterwards with a stack of free choice (Fig. 1).9
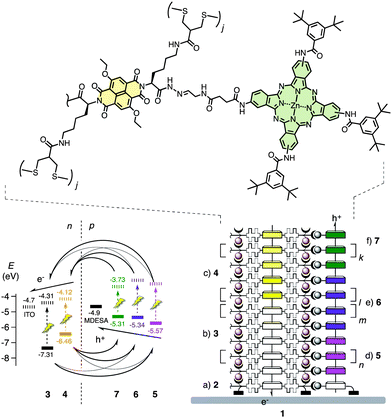 | ||
| Fig. 1 Design of a double-channel photosystem 1 with oriented antiparallel redox gradients composed of naphthalenediimides, phthalocyanines and porphyrins. Full structures of components 2–7 are shown in Scheme 1, their assembly by SOSIP-TSE is outlined in Scheme 2. HOMO (bold) and LUMO levels (dashed) are from the literature or calculated from cyclic voltammograms (−5.1 eV for Fc/Fc+). Charge transfer opportunities are based on feasibility and are confirmed partially, possible energy transfer is not shown. | ||
SOSIP has been achieved with naphthalenediimides (NDIs),6–9 perylenediimides (PDIs)6c and oligothiophenes,9b TSE with NDIs9a and PDIs.9b Here we report the compatibility of SOSIP-TSE with phthalocyanines1 and porphyrins,2 that are traditional components of an entirely different dimension that continue to play a major role in functional materials. Phthalocyanines and porphyrins are hole transporters with exceptionally strong absorption in the green and in the red, similar to chlorophyll. TSE along the electron-transporting NDI stacks obtained by SOSIP should thus afford double-channel architectures with good light harvesting properties (Fig. 1). Metalloporphyrins were of particular interest because of the varying HOMO energy levels and ease of metal exchange.2,10 Elaborating on this unique advantage, we report in situ access to up to three-component charge-transfer cascades along antiparallel two-component gradients in the NDI channel in photosystem 1, and show that such architectures are of interest to minimize charge recombination.11
The synthesis of initiator 2 and propagators 3 and 4 has been reported previously (Scheme 1).9a The synthesis of porphyrins 5 and 6 and phthalocyanine 7 required the introduction of two different types of substituents along the macrocyclic scaffold. Macrocyclizations with two different building blocks can be difficult and give low yields in the case of both porphyrins as well as phthalocyanines. We thus decided to use known macrocyclization procedures and introduce the two different substituents afterwards by sequential derivatization and isolation of the desired products from the obtained mixture.
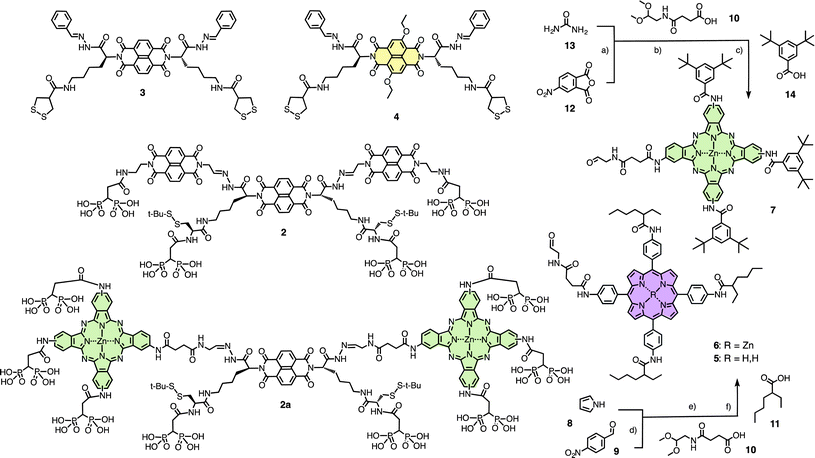 | ||
| Scheme 1 Structure of initiators 2 and 2a, and propagators 3 and 4, and synthesis and structure of stack exchangers 5–7. (a): (1) 12, 13, ZnCl2, 6 NH4.Mo7O24, nitrobenzene, 185 °C, 4 h, 83%, (2) Na2S, DMF, 65 °C, 16 h, 98%; (b) 10, HATU, collidine, DMF; (c): (1) 14, HATU, collidine, DMF, 2 h, rt, 32% (2 steps), (2) TMSBr, DCM, 2 h, rt (86%); (d): (1) 8, 9, propionic acid, 140 °C, 30 min, 17%; (2) SnCl2, HCl, 30 min, 77%; (e) 10, HATU, DIPEA, DMF, rt, 5 h; (f): (1) 11, HATU, DIPEA, DMF, 4 days, rt, 27% (2 steps), (2) TFA, DCM, 40 min, rt, 51%. | ||
Following this strategy, porphyrin 5 was synthesized from pyrrole 8 and nitrobenzaldehyde 9 under routine conditions.2 The nitro groups were reduced to amines. The tetraamino porphyrin was reacted first with one equivalent of ketal 10 and then, without purification, reacted with an excess of solubilizer 11. The desired product was isolated from the mixture of differently substituted tetraamido porphyrins in 27% yield over the two steps. Final deprotection of the aldehyde provided facile access to the target molecule 5. Zinc insertion into 5 to give 6 was possible, but was more conveniently done directly in the SOSIP-TSE architecture.
The same strategy was applied to prepare the phthalocyanine stack exchanger 7. Macrocyclization of anhydride 12 and urea 13 gave the previously reported phthalocyanines with four identical nitro substituents. Reduction of these peripheral nitro groups gave the tetraamino phthalocyanines. Analog to the synthesis of porphyrin 5, these amines were first reacted with one equivalent of ketal 10 and then with an excess of solubilizer 14 (several solubilizers had to be tested to identify 14 as the most powerful one). The desired phthalocyanine with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 substitution pattern could be easily isolated from the mixture of mainly 4:0, 3:1 and 2:2 substitution products. The target molecule 7 was obtained by final deprotection of the aldehyde.
:1 substitution pattern could be easily isolated from the mixture of mainly 4:0, 3:1 and 2:2 substitution products. The target molecule 7 was obtained by final deprotection of the aldehyde.
The phthalocyanine initiator 2a with giant phthalocyanine templates for stack exchange was prepared analogous to initiator 2. In brief, the tetraamino phthalocyanines were reacted first with one equivalent of ketal 10 and then with the benzyl protected diphosphonate “feet” that have been reported previously.9 The desired phthalocyanine with a 3:1 substitution pattern was isolated from the mixture of products, and the aldehyde was deprotected. In situ hydrazone formation with the previously reported NDI templates9 gave the target molecule 2a. All details on organic multistep synthesis can be found in the ESI.†
Initiators 2 and 2a contain four to eight diphosphonate “feet” to bind to ITO surfaces. The covered surfaces were characterized by cyclic voltammetry (CV) and then activated with DTT. SOSIP was initiated by molecular recognition of propagators on the surface of 15 (Scheme 2). Ring-opening disulfide exchange polymerization of the propagator 4 afforded NDI stack 16. After SOSIP, the benzaldehyde hydrazones were cleaved by the treatment with hydroxylamine to afford photosystem 17 with reactive hydrazide pores next to the NDI stacks.9 These holes were then filled with phthalocyanine 7 to give heterojunction photosystem 18 (Fig. 2a).
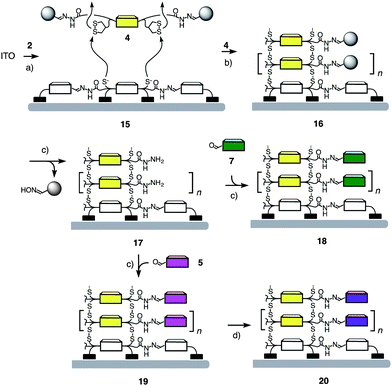 | ||
| Scheme 2 SOSIP-TSE protocol. (a) Initiator deposition and activation: (1) 3 mM 2 (or 1 mM 2a), DMSO, 1–2 days, rt; (2) 120 °C, 1 h; (3) 20 mM DTT, 100 mM aq. NH4HCO3, 1 h, rt. (b) SOSIP: 7 mM 4, CHCl3/MeOH 1:1, 100 mM DIPEA, 24 h, rt. (c) Stack exchange: (1) 1 M NH2OH·HCl, pH 3, 1 day, 40 °C; (2) 10 mM 7 (or 5), 10% AcOH, DMSO. (d) Zn(OAc)2, MeOH, 10 min. | ||
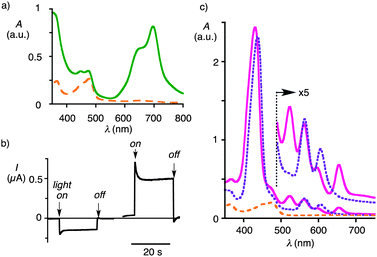 | ||
| Fig. 2 (a) Absorption spectra of SOSIP photosystems prepared from the initiator 2a before (16a, dashed) and after TSE with 7 (18a, solid line). (b) Photocurrent generation by photosystems 18a (left) and 18 (right) upon irradiation with a solar simulator (42 mW cm−2) at 0 V vs. Ag/AgCl. (c) Absorption spectra of photosystems prepared from the initiator 2 before (16, dashed) and after TSE with 5 (19, solid) followed by a Zn insertion (20, dotted line). | ||
The absorption spectra of the film showed surprisingly weak absorbance of phthalocyanines near 700 nm with characteristic features of H-aggregates. Because of the apparently significant hypochromism of phthalocyanines, the yield of stack exchange was estimated from the absorption spectra after dissolving the SOSIP film using excess mercaptoethanol (Fig. S16b†). The obtained yields (quantitative with initiator 2; 48% with 2a) indicated that the stack exchange is insensitive to the mismatch in size of template and stack. Poorer yield for stack exchange with the initiator 2a could be attributed to less organized SOSIP due to large empty areas available next to the growing polymers.
The photocurrent measurements were done using the photosystem as a working electrode together with a Pt counter electrode, an Ag/AgCl reference electrode and disodium methoxyaniline N,N-bis(ethyl sulfonate) (MDESA)12 as a mobile electron carrier. Irradiated with a solar simulator, photosystem 18 grown from initiator 2 generated modest photocurrent while that with 2a gave negative photocurrent (Fig. 2b). These results indicated that the NDI template as in initiator 2 is essential for the function of photosystems by acting as a hole blocker on the electrode surface. Thus, subsequent studies were conducted only using the initiator 2.
SOSIP photosystems with porphyrins (19) were similarly prepared using the aldehyde 5 instead of 7. Immersion of 19 in a solution of Zn(OAc)2 resulted in a quantitative insertion of Zn2+ in the porphyrin core to give photosystem 20.10 Other metals, such as Pd2+ and Cu2+ could be similarly inserted in the porphyrin core. However, the photocurrent generation by the resulting photosystems was clearly inferior to that with free-base porphyrin 19 or Zn-porphyrin 20.
With these hole transporters in hand, construction of double-channel photosystems with antiparallel redox gradients was straightforward. Namely, successive SOSIP of propagators 3 and then 4 on the surface of an ITO electrode coated with the activated initiator 2 afforded NDI stacks with two-component gradients that drive the electrons toward the ITO surface (Fig. 1).9 After TSE with porphyrin 5, the redox gradients were introduced in the hole-conducting pathway of photosystem 21 either by partial Zn2+ insertion (22) or by partial stack exchange with phthalocyanine 7 (23, Fig. 3). The three-component cascade 1 was made by the combination of the two. As in the previous report,9a the effect of the redox gradients was evaluated by bimolecular recombination efficiency ηBR, which can be derived from the dependence of photocurrent on the irradiation power. As expected, about 0.2 eV of differences in HOMO energy levels of both porphyrin-Zn porphyrin (5 to 6) and porphyrin-Zn phthalocyanine (5 to 7) gradient were sufficient to lower the ηBR by driving holes to the opposite direction to that of electrons (Fig. 1 and 3d). However, the three-component gradient in photosystem 1 did not give rise to a meaningful improvement. This result could be ascribed to the too small difference in HOMO energy levels of Zn-porphyrin 6 and Zn-phthalocyanine 7 (Fig. 1).13 Overall, the application of antiparallel redox gradients in photosystems 1, 22 and 23 gave recombination efficiencies of ηBR = 35–36%, whereas photosystem 21 with only one redox gradient gave ηBR = 48%. These results were in agreement with previous findings with NDI-based photosystems. In that case, antiparallel two-component gradients gave ηBR = 22%, whereas gradient-free controls gave ηBR = 50%.9a Nearly the same ηBR found with single-gradient photosystem 21 is consistent with the previous observation5b and confirmed that one gradient is insufficient to minimize charge recombination. The decrease in ηBR with double-gradient photosystems 1, 22 and 23, although less pronounced than that with NDI-based photosystems, confirmed the general validity of the idea that antiparallel gradients can help minimizing charge recombination by driving holes and electrons in opposite directions.
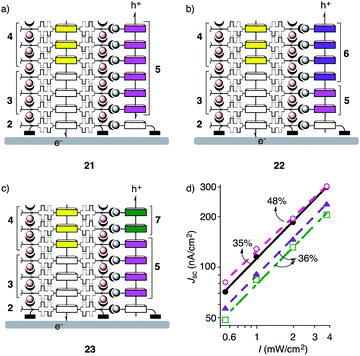 | ||
| Fig. 3 (a–c) Schematic structure of multicomponent systems. (d) Dependence of short circuit current densities (JSC) on light intensities (I) for SOSIP-TSE photosystems 1 (●), 21 (○), 22 (▲) and 23 (□) with bimolecular charge recombination efficiencies ηBR indicated in %. | ||
Conclusions
In summary, we have demonstrated the compatibility of SOSIP-TSE methodology with well-established chromophores from classical photosystems, i.e., porphyrins and phthalocyanines. Tolerance of TSE to the template-stack mismatch in size is in sharp contrast to the extreme sensitivity of SOSIP process to the minor structural variations.9a,8,6b Such properties are advantageous for the construction of highly complex but ordered multicomponent architectures. On the functional level, the observed positive effect of the NDI template as a hole blocker implies similarly beneficial effects to an electron-blocking layer. This could be achieved easily by SOSIP of more electron-rich propagators, for example diamino substituted naphthalenediimides (blue NDIs), as a third component of the electron-transporting cascade.6Toward the installation of multicomponent redox gradients, we report, for the first time, the assembly of a hole-conducting cascade composed of three components (or even four components if we also count the NDI hole blocker in the template). Further elaboration would be possible by using other metallated porphyrins. Overall however, the effect of the redox gradient was disappointingly low in this system. This might be caused by the poor charge separation between porphyrins/phthalocyanines and NDIs evidenced in action spectra (Fig. S16†). Thus, to harness the potential of these chromophores, it is imperative to find and incorporate better electron acceptors into the SOSIP-TSE photosystems. The remarkable tolerance found for templated stack exchange suggests that there should be room for a third channel, or even more. Studies toward this goal are ongoing; preliminary results with triple channel architectures are encouraging.
Acknowledgements
We thank D.-H. Tran, Z. Truan and A. Cherix for assistance in synthesis, D. Jeannerat, A. Pinto and S. Grass for NMR measurements, the Sciences Mass Spectrometry (SMS) platform for mass spectrometry services, and the University of Geneva, the European Research Council (ERC Advanced Investigator), the National Centre of Competence in Research (NCCR) Chemical Biology and the Swiss NSF for financial support. G. S. is a Curie fellow.Notes and references
- (a) G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768–6816 CrossRef CAS; (b) M. V. Martínez-Díaz, G. de la Torre and T. Torres, Chem. Commun., 2010, 46, 7090–7108 RSC; (c) M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 759–792 CrossRef CAS; (d) E. Göransson, J. Boixel, J. Fortage, D. Jacquemin, H.-C. Becker, E. Blart, L. Hammarström and F. Odobel, Inorg. Chem., 2012, 51, 11500–11512 CrossRef; (e) A. Membrino, M. Paramasivam, S. Cogoi, J. Alzeer, N. W. Luedtke and L. E. Xodo, Chem. Commun., 2010, 46, 625–627 RSC; (f) F. Yang, M. Shtein and S. Forrest, Nat. Mater., 2005, 4, 37–41 CrossRef CAS.
- (a) H. Jia, B. Schmid, S. X. Liu, M. Jaggi, P. Monbaron, S. V. Bhosale, S. Rivadehi, S. J. Langford, L. Sanguinet, E. Levillain, M. E. El-Khouly, Y. Morita, S. Fukuzumi and S. Decurtins, ChemPhysChem, 2012, 13, 3370–3382 CrossRef CAS; (b) R. Charvet, Y. Yamamoto, T. Sasaki, J. Kim, K. Kato, M. Takata, A. Saeki, S. Seki and T. Aida, J. Am. Chem. Soc., 2012, 134, 2524–2527 CrossRef CAS; (c) S. Fukuzumi, K. Ohkubo, F. D'Souza and J. L. Sessler, Chem. Commun., 2012, 48, 9801–9815 RSC; (d) T. J. Bandy, A. Brewer, J. R. Burns, G. Marth, T. Nguyen and E. Stulz, Chem. Soc. Rev., 2011, 40, 138–148 RSC; (e) L. P. Hernandez-Eguia, R. J. Brea, L. Castedo, P. Ballester and J. R. Granja, Chem.–Eur. J., 2011, 17, 1220–1229 CrossRef CAS; (f) M. C. O'Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. Claridge, A. Saywell, M. O. Blunt, J. N. O'Shea, P. H. Beton, M. Malfois and H. L. Anderson, Nature, 2011, 469, 72–75 CrossRef CAS; (g) A. Yella, H. W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS; (h) A. Kira, T. Umeyama, Y. Matano, K. Yoshida, S. Isoda, J. K. Park, D. Kim and H. Imahori, J. Am. Chem. Soc., 2009, 131, 3198–3200 CrossRef CAS; (i) J. Wiberg, L. Guo, K. Pettersson, D. Nilsson, T. Ljungdahl, J. Mårtensson and B. Albinsson, J. Am. Chem. Soc., 2006, 129, 155–163 CrossRef; (j) P. Ballester, A. Costa, P. M. Deyà, A. Frontera, R. M. Gomila, A. I. Oliva, J. K. M. Sanders and C. A. Hunter, J. Org. Chem., 2005, 70, 6616–6622 CrossRef CAS; (k) D. M. Guldi, I. Zilbermann, G. Anderson, A. Li, D. Balbinot, N. Jux, M. Hatzimarinaki, A. Hirsch and M. Prato, Chem. Commun., 2004, 726–727 RSC; (l) D. Kim and A. Osuka, Acc. Chem. Res., 2004, 37, 735–745 CrossRef CAS; (m) T. van der Boom, R. T. Hayes, Y. Zhao, P. J. Bushard, E. A. Weiss and M. R. Wasielewski, J. Am. Chem. Soc., 2002, 124, 9582–9590 CrossRef CAS; (n) S. Matile, N. Berova, K. Nakanishi, J. Fleischhauer and R. W. Woody, J. Am. Chem. Soc., 1996, 118, 5198–5206 CrossRef CAS.
- (a) F. Würthner and K. Meerholz, Chem.–Eur. J., 2010, 16, 9366–9373 CrossRef; (b) D. M. Bassani, L. Jonusauskaite, A. Lavie-Cambot, N. D. McClenaghan, J.-L. Pozzo, D. Ray and G. Vives, Coord. Chem. Rev., 2010, 254, 2429–2445 CrossRef CAS; (c) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS; (d) T. W. Holcombe, J. E. Norton, J. Rivnay, C. H. Woo, L. Goris, C. Piliego, G. Griffini, A. Sellinger, J. L. Brèdas, A. Salleo and J. M. Frèchet, J. Am. Chem. Soc., 2011, 133, 12106–12114 CrossRef CAS; (e) R. Fitzner, E. Reinold, A. Mishra, E. Mena-Osteritz, H. Ziehlke, C. Körner, K. Leo, M. Riede, M. Weil, O. Tsaryova, A. Weiß, C. Uhrich, M. Pfeiffer and P. Bäuerle, Adv. Funct. Mater., 2011, 21, 897–910 CrossRef CAS; (f) R. J. Kumar, J. M. MacDonald, T. B. Singh, L. J. Waddington and A. B. Holmes, J. Am. Chem. Soc., 2011, 133, 8564–8573 CrossRef CAS; (g) J. L. Delgado, P.-A. Bouit, S. Filippone, M. A. Herranz and N. Martín, Chem. Commun., 2010, 46, 4853–4865 RSC; (h) R. Bhosale, J. Míšek, N. Sakai and S. Matile, Chem. Soc. Rev., 2010, 39, 138–149 RSC; (i) J. Roncali, Acc. Chem. Res., 2009, 42, 1719–1730 CrossRef CAS.
- (a) A. Perez-Velasco, V. Gorteau and S. Matile, Angew. Chem., Int. Ed., 2008, 47, 921–923 CrossRef CAS; (b) S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS.
- (a) N. Sakai, A. L. Sisson, T. Bürgi and S. Matile, J. Am. Chem. Soc., 2007, 129, 15758–15759 CrossRef CAS; (b) N. Sakai, R. Bhosale, D. Emery, J. Mareda and S. Matile, J. Am. Chem. Soc., 2010, 132, 6923–6925 CrossRef CAS; (c) R. Bhosale, R. S. K. Kishore, V. Ravikumar, O. Kel, E. Vauthey, N. Sakai and S. Matile, Chem. Sci., 2010, 1, 357–368 RSC.
- (a) N. Sakai, M. Lista, O. Kel, S. Sakurai, D. Emery, J. Mareda, E. Vauthey and S. Matile, J. Am. Chem. Soc., 2011, 133, 15224–15227 CrossRef CAS; (b) M. Lista, J. Areephong, N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 15228–15230 CrossRef CAS; (c) P. Charbonnaz, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1492–1496 RSC.
- E.-K. Bang, M. Lista, G. Sforazzini, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1752–1763 RSC.
- E. Orentas, M. Lista, N.-T. Lin, N. Sakai and S. Matile, Nat. Chem., 2012, 4, 746–750 CrossRef CAS.
- (a) N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 18542–18545 CrossRef CAS; (b) J. Areephong, E. Orentas, N. Sakai and S. Matile, Chem. Commun., 2012, 48, 10618–10620 RSC.
- (a) A. Giraudeau, F.-R. F. Fan and A. J. Bard, J. Am. Chem. Soc., 1980, 102, 5137–5142 CrossRef CAS; (b) T. D. Santos, A. Morandeira, S. Koops, A. J. Mozer, G. Tsekouras, Y. Dong, P. Wagner, G. Wallace, J. C. Earles, K. C. Gordon, D. Officer and J. R. Durrant, J. Phys. Chem. C, 2010, 114, 3276–3279 CrossRef; (c) U. Weiler, T. Mayer, W. Jaegermann, C. Kelting, D. Schlettwein, S. Makarov and D. Wöhrle, J. Phys. Chem. B, 2004, 108, 19398–19403 CrossRef CAS; (d) R. Chitta, L. M. Rogers, A. Wanklyn, P. A. Karr, P. K. Kahol, M. E. Zandler and F. D'Souza, Inorg. Chem., 2004, 43, 6969–6978 CrossRef CAS; (e) M. Gervaldo, F. Fungo, E. N. Durantini, J. J. Silber, L. Sereno and L. Otero, J. Phys. Chem. B, 2005, 109, 20953–20962 CrossRef CAS.
- Architectures with oriented multicomponent antiparallel redox gradients in co-axial hole- and electron-transporting channels have been referred to as OMARG-SHJs (SHJ, supramolecular n/p-heterojunctions)5b,9a.
- Y. Kim, S. Keller, J. Krueger, E. Yonemoto, G. Saupe and T. E. Mallouk, J. Phys. Chem. B, 1997, 101, 2491–2500 CrossRef CAS.
- The difference in HOMO levels of Zn-phthalocyanines and Zn-porphyrins is estimated to be about 30 mV, see ref. 10c.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures. See DOI: 10.1039/c3sc00041a |
| This journal is © The Royal Society of Chemistry 2013 |