 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benzyl bispidine as an efficient replacement for (−)-sparteine in ring opening polymerisation†
Richard
Todd
a,
Gabriel
Rubio
ab,
Daniel J.
Hall
a,
Sarah
Tempelaar
a and
Andrew P.
Dove
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: a.p.dove@warwick.ac.uk; Tel: +44 (0)24 7652 4107
bUniversity of Barcelona, Martí i Franquès Street, 1 08028, Barcelona, Spain
First published on 12th December 2012
Abstract
The synthesis and application of a dibenzyl-functionalized bispidine, in combination with 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea (TU) co-catalyst, has been demonstrated to be an excellent catalyst for the controlled ring-opening polymerisation (ROP) of lactide and cyclic carbonate monomers. Notably, the polymerisation proceeds with negligible transesterification or epimerisation, with the polymerisation of stereopure L-lactide affording highly crystalline poly(lactide) with a Tm of 156 °C. ROP of racemic lactide results in the observation of a modest degree of stereocontrol such that the probability of isotactic enchainment, Pm = 0.74. Comparison of a range of alternative hydrogen bond donor co-catalysts revealed that TU displayed the highest polymerisation rates in combination with the dibenzyl-functionalized bispidine.
Introduction
The last decade has led to many advances in organocatalytic ring-opening polymerisation (ROP) of several cyclic monomers, most commonly including esters and carbonates.1–8 On account of their facile access, commercial availability and efficient nature, organic catalysts have become a valuable addition to this field. While several simple species have been reported to mediate these processes, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), triazabicyclodecene (TBD) and N-heterocyclic carbenes are amongst the most active,9–14 the bifunctional catalyst system containing both a thiourea and tertiary amine (Fig. 1), has been reported to proceed with an almost absence of transesterification and epimerization side reactions, and thus provides a critical method for the production of precisely defined polymers. The high selectivity towards polymerisation in preference to deleterious side reactions is attributed to the supramolecular recognition (and concurrent activation) of the cyclic ester monomers in preference to the linear esters by the thiourea moiety.15,16 Critically however, it was demonstrated that the thiourea and the amine need not be conjoined and in turn this has led to the discovery of more active analogues, most notably the combination of 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea (TU) and (−)-sparteine, a naturally occurring alkaloid, which finds the optimum balance between activity and selectivity.16 The increased rate (25 fold increase) can be assigned to the two tertiary amines of (−)-sparteine being forced into close proximity as a consequence of the fixed rigid backbone with the lone pairs being oriented towards each other in such a way to promote hydrogen bonding. Concurrently, the weakly basic nature of (−)-sparteine (pKa = 21.66),17 maintains the high selectivity towards ROP vs. transesterification and epimerisation. This concept has since been extended to a wide range of hydrogen bond acceptors and donors that maintain the simultaneous activation of both the carbonyl group of the monomer and the initiating/propagating alcohol.18–22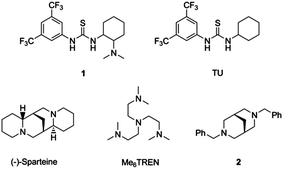 | ||
| Fig. 1 Ring opening-polymerisation catalysts. | ||
In addition to its uses in ROP, (−)-sparteine has a number of other uses such as a chiral ligand in asymmetric deprotonations, substitutions and metalations.23–26 Despite this widespread use and utility, (−)-sparteine has become increasingly difficult to acquire through commercial sources. Several potential routes to obtain (−)-sparteine exist including isolation from the abundant Scotch Broom (Cytisus scoparius),27 or total synthesis via a multistep route that has varying overall yields.28,29 Furthermore, the derivation of synthetic replacements may enable more highly active and stereoselective analogues to be realised.
These challenges to obtaining (−)-sparteine have led to the investigation of synthetically simple analogues. Aided by computational modelling, Hedrick and co-workers recently identified several commercially available tertiary amines that displayed similar nitrogen–nitrogen spacing (∼3 Å) and lone pair orientations to (−)-sparteine.30 Of the tertiary amines selected, tris[2-dimethylamino)ethyl]amine (Me6TREN) and 1,4,7-trimethyl-1,4,7-triazacyclononane (TACN), in combination with TU, proved the most effective co-catalysts in the ROP of lactide. While TACN displayed comparable polymerization kinetics to (−)-sparteine, transesterification was noted to occur rapidly upon complete conversion of the monomer. Application of Me6TREN led to better polymerisation control albeit at a slightly longer polymerisation time.
We hypothesised that a closer structural analogue to (−)-sparteine would lead to more comparable behaviour in ROP, and hence targeted the preparation of analogous bispidines. Herein we report the improved synthesis of a dibenzyl-functional bispidine, chosen for its identical backbone and similar basicity (pKa = 21.25) to (−)-sparteine in addition to its apparent ease of synthesis,17 and explore its activity in the ROP of lactide, comparing directly to that of (−)-sparteine and Me6TREN. Extension of the application of this species with a range of co-catalysts and other monomer feedstocks is also demonstrated.
Results and discussion
Bispidine synthesis and lactide ROP
The synthesis of benzyl bispidine has been reported previously by two different synthetic routes. While one route involves a double Mannich reaction of 1-benzyl-4-piperidone followed by a Wolff–Kishner reduction with hydrazine,31 on account of the broader substrate versatility, we preferred to apply the alternative four step synthetic route reported by Gogoll et al. starting from dimethyl malonate, 3 (Scheme 1).17,32 Following the Knoevenagel condensation of 3 with paraformaldehyde, reduction of the resulting adduct using LiAlH4 yielded the tetra-alcohol species, 5. Iodination of 5 as previously reported proved to be problematic on large scales and hence bromination with phosphorous tribromide was undertaken.33 The crude product could be readily purified by passing through a silica plug to obtain 6b as a white solid in 61% yield. Reaction of 6b with excess benzyl amine enabled the isolation of pure bispidine, 2, after flash column chromatography using a selective gradient solvent system.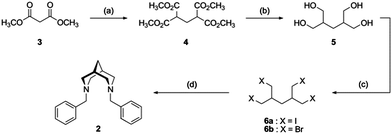 | ||
| Scheme 1 Benzyl bispidine synthesis. (a) Paraformaldehyde, KOH, 95 °C; (b) LiAlH4, THF; (c) 6a: I2, Pred, 120 °C; 6b: PBr3, 100 °C; (d) benzylamine, toluene, reflux. | ||
The ROP of L-lactide, LA, using 10 mol% TU and 5 mol% 2, was studied in CDCl3 ([LA]0 = 0.7 M) at ambient temperature using 1-phenylethanol as the initiator at a monomer-to-initiator ratio of 50 ([M]0/[I]0 = 50). Monitoring the polymerisation by 1H NMR spectroscopy revealed that it had reached 87% monomer conversion after only 81 min. Analysis of the resultant polymer by gel-permeation chromatography (GPC) revealed a number average molecular weight (Mn) of 13.4 kg mol−1 with a narrow dispersity (ĐM = 1.08). Under the same conditions, polymerisation of L-LA with (−)-sparteine and Me6TREN in place of 2 reached 93% and 92% monomer conversion after 66 and 450 min respectively with similar molecular parameters for the resulting PLA observed by GPC analysis (Table 1). To more precisely compare catalyst reactivities, kinetic studies of LA ROP for each catalyst system were investigated in triplicate (Fig. 2). First order kinetic plots of ln([M]0/[M]t) against time reveal a linear correlation, with apparent rate constants (Kapp) calculated as 1.35 h−1, 2.53 h−1 and 0.38 h−1 for 2, (−)-sparteine and Me6TREN in combination with TU respectively. Under these conditions, while the bispidine catalyses lactide ROP at a lower rate than (−)-sparteine, the polymerisation occurs almost four times faster then when Me6TREN is applied.
| Catalysta | Conversionb (%) | Time (min) | M n (g mol−1) | Đ M | K app (h−1) |
|---|---|---|---|---|---|
| a Conditions: 1 ml of CDCl3 at room temperature; [L-LA] = 0.7 M; 1-phenylethanol as initiator; 5 mol% tertiary amine; 10 mol% of TU; [M]0/[I]0 = 50. b Determined by 1H NMR spectroscopy. c Determined by GPC analysis in CHCl3. | |||||
| (−)-Sparteine | 93 | 66 | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 900 |
1.08 | 2.53 |
| 2 | 87 | 81 | 13400 |
1.08 | 1.35 |
| Me6TREN | 92 | 450 | 12100 |
1.09 | 0.38 |
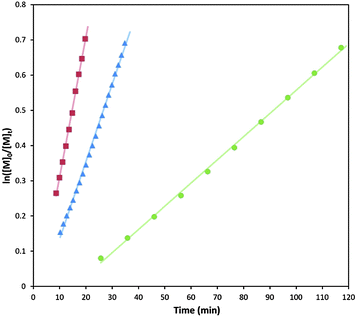 | ||
| Fig. 2 Semi-log kinetic plot for the ROP of L-LA using TU in combination with a tertiary amine cocatalyst. (Red = (−)-sparteine; blue = 2; green = Me6TREN). | ||
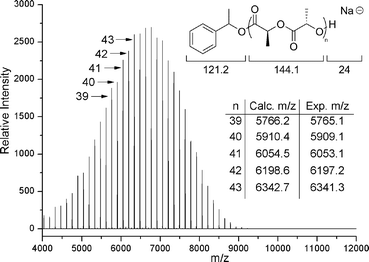
The controlled nature of the benzyl bispidine-catalysed polymerisation was further verified by the linear correlation between Mn values obtained by GPC and the monomer conversion determined by 1H NMR spectroscopy (Fig. 3a) and the [M]/[I] ratio (Fig. 3b). The resultant PLAs all displayed narrow dispersities (1.18–1.03, Table 2) which indicates that minimal transesterification is occurring. Further analysis of the resultant polymers by matrix-assisted laser desorption ionisation time of flight mass spectrometry (MALDI-ToF MS) revealed only single distributions with spacings of 144 m/z between neighbouring peaks (Fig. 4). The absence of a second distribution with spacings of 72 m/z is consistent with the absence of transesterification side reactions.
![(a) Number-average molecular weight (Mn) against % monomer conversion and (b) number-average molecular weight (Mn) and dispersity (ĐM) against initial monomer-to-initiator ratio ([M]0/[I]0) for the ROP of l-LA using TU and 2.](/image/article/2013/SC/c2sc22053a/c2sc22053a-f3.gif) | ||
| Fig. 3 (a) Number-average molecular weight (Mn) against % monomer conversion and (b) number-average molecular weight (Mn) and dispersity (ĐM) against initial monomer-to-initiator ratio ([M]0/[I]0) for the ROP of L-LA using TU and 2. | ||
| [M]/[I] | Conversionb (%) | Time (min) | M n (g mol−1) | Đ M |
|---|---|---|---|---|
| a Conditions: CDCl3; room temperature; [L-LA] = 0.7 M; 1-phenylethanol as initiator; 5 mol% of 2; 10 mol% of TU. b Determined by 1H NMR spectroscopy. c Determined by GPC analysis in CHCl3. | ||||
| 10 | 90 | 21 | 3280 | 1.18 |
| 20 | 94 | 43 | 6050 | 1.09 |
| 50 | 92 | 75 | 13200 |
1.08 |
| 100 | 89 | 137 | 26150 |
1.06 |
| 250 | 90 | 366 | 61600 |
1.03 |
Thermal analysis of PLLAs with a degree of polymerisation (DP) of 100 synthesised using TU with both 2 and (−)-sparteine observed a melting temperature, Tm, of 156 and 158 °C respectively. These match the previously reported melting points for isotactic PLLA prepared using (−)-sparteine.34,35 In combination with the observation of a singlet resonance in both the homonuclear decoupled 1H NMR and 13C NMR spectra of PLLA synthesised in this manner, these observations confirm the absence of stereo errors, and hence an absence of epimerisation, with identical levels resulting from both (−)-sparteine and 2 catalysed polymerisations.36–39
ROP of rac-lactide was undertaken to determine if any stereoselectivity would be observed using the catalyst combination of TU and 2. Despite the absence of stereogenic centres in 2, unlike (−)-sparteine, analysis of a DP100 poly(rac-lactide) by homonuclear decoupled 1H NMR spectroscopy enabled the calculation of the probability of isotactic enchainment, Pm, to be 0.74, clearly demonstrating a preference to produce isotactic PLA from the racemic monomer mixture. This is comparable to that observed for rac-LA ROP with (−)-sparteine–TU which resulted in a Pm = 0.74 (literature value: Pm = 0.77).16
Variation of the hydrogen bond donor co-catalyst
Several hydrogen bond donor alternatives to the thiourea co-catalyst (TU) have been reported over recent years that are successfully used in conjunction with (−)-sparteine (Fig. 5).18–22 In order to assess the efficiency of these co-catalysts in conjunction with the dibenzyl-functional bispidine, 2, in comparison to the initially reported system, we studied their activity for the ROP of L-lactide.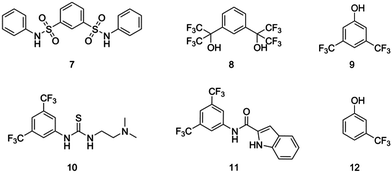 | ||
| Fig. 5 Hydrogen bond donor co-catalysts. | ||
The ROP of L-lactide, LA, using 5 mol% 2 with 10 mol% hydrogen bond donor co-catalyst was again studied in CDCl3 ([LA]0 = 0.7 M) at ambient temperature using 1-phenylethanol as the initiator at a monomer-to-initiator ratio of 50 ([M]0/[I]0 = 50). The limited solubility of 11 in chloroform required its application at the lower molar ratio of 5 mol% (in preference to the standard 10 mol%). To provide an accurate comparison, polymerisations using co-catalyst TU were also undertaken at 5 mol% loading. Kinetic studies for each catalyst system were investigated in triplicate by monitoring monomer conversion against time using 1H NMR spectroscopy. First order kinetic plots of ln([M]0/[M]t) against time (Fig. 6) enabled the determination of Kapp values (Table 3). The tested catalysts demonstrated a broad range of activities, with Kapp values ranging from 1.35 h−1 to 0.03 h−1. While TU at 10 mol% loading resulted in the most rapid polymerisation, the alternative co-catalysts were observed to display much lower Kapp values such that the second most active co-catalyst is m-trifluoromethylphenol, 12 with a Kapp approximately an order of magnitude lower than that observed for TU. Notably the amide catalyst 11 (5 mol% loading) remained highly active with the measured Kapp value approximately 6 times lower than that TU at the reduced 5 mol% loading. Polymerisation using co-catalyst 7 displayed extremely low reaction rate under these conditions, with <5% monomer conversion being observed after one week.
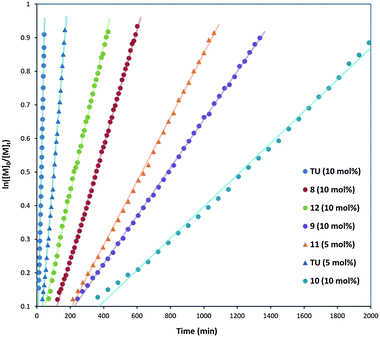 | ||
| Fig. 6 Kinetic plots resulting from variation in hydrogen bond donor. | ||
| Co-Catalyst | [M]/[I] | Conversionc (%) | Time (min) | M n (g mol−1) | Đ M | K app (h−1) |
|---|---|---|---|---|---|---|
| a Conditions: 1 ml of CDCl3 at room temperature; [L-LA] = 0.7 M; 5 mol% 2; 10 mol% co-catalyst; 1-phenylethanol as initiator. b Conditions: 1 ml of CDCl3 at room temperature; [L-LA] = 0.7 M; 5 mol% 2; 5 mol% co-catalyst; 1-phenylethanol as initiator. c Determined by 1H NMR spectroscopy. d Determined by GPC analysis in CHCl3. | ||||||
| TUa | 50 | 86 | 75 | 13500 |
1.08 | 1.354 ± 0.053 |
| TUb | 50 | 90 | 350 | 12750 |
1.07 | 0.391 ± 0.027 |
| 8a | 50 | 86 | 1220 | 8450 | 1.06 | 0.103 ± 0.001 |
| 9a | 50 | 91 | 3240 | 9090 | 1.11 | 0.044 ± 0.002 |
| 10a | 50 | 86 | 4200 | 10300 |
1.08 | 0.029 ± 0.001 |
| 11b | 50 | 92 | 2710 | 11900 |
1.08 | 0.063 ± 0.007 |
| 12a | 50 | 93 | 1470 | 4010 | 1.20 | 0.138 ± 0.007 |
In each case, analysis of the molecular parameters of the resultant polymers revealed low dispersity values which suggests that the polymerisations were well controlled (Table 3). Notably however, co-catalyst 12 resulted in PLLA with lower molecular weight than predicted from the [M]0/[I]0 ratio. MALDI-ToF MS analysis of this polymer revealed two distinct distributions, both displaying the expected 144 m/z spacing for the monomer repeat unit. A molecular weight difference of ∼40 m/z was observed between the distributions. One of these distributions revealed values that correspond to sodium-charged polymer chains initiated from 1-phenylethanol as expected, however the other distribution showed values revealed values that correspond to sodium-charged polymer chains initiated from the co-catalyst 12, which possibly contributes to the increased observed rate of polymerisation. In the case of 9, in which two CF3 substituents are present on the phenol ring, co-catalyst initiation was not observed which suggests that the phenol in 9 is appropriately activated to only undertake hydrogen bonding and not serve as a source of initiation in the polymerisation.
Interestingly, 1H NMR spectroscopic analysis of the polymer produced using 2/8 as co-catalysts for the ROP of L-LA revealed a number of overlapping quartets in the range of 5.10–5.25 ppm, consistent with the epimerisation of the monomer during the polymerisation. Despite the previous report of the ROP of L-LA using (−)-sparteine/8 proceeding in the absence of epimerisation,22 in our hands, comparable 1H NMR spectra of the resultant polymers were also obtained for this system.
ROP of cyclic carbonates
To further investigate the versatility of the 2–TU binary catalyst system, polymerisations were undertaken of the 6-membered cyclic carbonates, trimethylene carbonate (TMC) and 5-methyl-5-allyloxycarbonyl-1,3-dioxan-2-one (MAC) of which the (−)-sparteine–TU system has previously demonstrated well controlled homopolymerisation.40–42 Polymerisations were carried out under conditions published in literature, using both 2 and (−)-sparteine in combination with TU as catalysts with a target [M]0/[I]0 = 50 for TMC and 20 for MAC.The ROP of TMC was performed using 5 mol% of 2 or (−)-sparteine with 5 mol% TU. After ca. 70 h 1H NMR spectroscopic analysis of the polymerisation mixtures revealed monomer conversions >90% (Table 4). GPC analysis of the resultant poly(TMC)s showed similar Mn values resulted from polymerisations using both catalysts with narrow dispersities observed. MALDI-ToF analysis of the poly(TMC) synthesised using 2–TU revealed a single distribution having the desired spacing of 102 m/z. No secondary distributions were observed in the MALDI-ToF spectra.
| Monomer | Catalyst | [M]0/[I]0 | Conversionc (%) | Time (min) | M n (g mol−1) | Đ M |
|---|---|---|---|---|---|---|
| a Conditions: 0.5 ml of CDCl3 at room temperature; [TMC] = 2.0 M; 5 mol% tertiary amine; 5 mol% TU; benzyl alcohol as initiator. b Conditions: 0.5 ml of CDCl3 at room temperature; [MAC] = 0.5 M; 5 mol% tertiary amine; 10 mol% TU; benzyl alcohol as initiator. c Determined by 1H NMR spectroscopy. d Determined by GPC analysis in CHCl3. e Determined by GPC analysis in DMF. | ||||||
| TMCa | (−)-Sparteine | 50 | 92 | 4280 | 9100d | 1.03d |
| TMCa | 2 | 50 | 90 | 4750 | 9100d | 1.03d |
| MACb | (−)-Sparteine | 20 | 90 | 400 | 6400e | 1.11e |
| MACb | 2 | 20 | 90 | 410 | 6200e | 1.10e |
| MACb | Me6TREN | 20 | 89 | 45 days | 6100e | 1.13e |
The polymerisations of MAC were undertaken using 5 mol% of 2 or (−)-sparteine with 10 mol% TU, as recently reported.41 Targeting a DP of 20, polymerisations catalysed by TU in combination with 2 or (−)-sparteine achieved 90% monomer conversion after 410 and 400 min respectively. Notably these results demonstrate the comparable activity of both bispidine co-catalysts for the ROP of cyclic carbonates. In both cases, GPC analysis revealed that the molecular weights and dispersities for the resulting poly(MAC) were within error and MALDI-ToF MS analysis revealed only the desired single distribution with the predicted spacing of 200 m/z that corresponds to the molecular weight of the repeat unit. Interestingly, the application of 10 mol% TU with 5 mol% Me6TREN revealed a slower polymerization such that 89% conversion was recorded after 45 days. GPC analysis of the resultant polymer revealed molecular weights and dispersities in close agreement to (−)-sparteine and 2.
Conclusions
In this paper we demonstrate that, in conjunction with a hydrogen bond donor co-catalyst, the dibenzyl-functional bispidine, 2, is an excellent replacement for (−)-sparteine in the ROP of lactide, producing well controlled polymers in the absence of observable transesterification and epimerisation. Additionally the ROP of rac-lactide revealed similar stereoselectivities for 2 and (−)-sparteine, with a Pm of 0.74 calculated for both catalyst systems. Optimisation of the co-catalyst revealed that 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea (TU) resulted in the highest observed polymerisation rates. In addition, the ROP of cyclic carbonates with 2 was explored achieving rates almost identical to those observed using (−)-sparteine whilst the control over the molecular weight and dispersity was maintained. In conclusion, benzyl bispidine has proven to be a versatile catalyst and an excellent replacement for (−)-sparteine.Acknowledgements
The Research Councils UK (RCUK) are acknowledged for funding a fellowship to A.P.D. We gratefully acknowledge financial support from EPSRC for funding a DTA studentship (R.T.) and a postdoctoral fellowship to D.J.H. (EP/H034420/1) as well as the purchase of the Bruker Ultraflex MALDI-ToF MS instrument. The ERASMUS programme is acknowledged for funding to support G.R. The GPC and thermal analysis equipment used in this research was obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). Purac are gratefully acknowledged for the kind donation of lactide monomers, Aldrich Materials Science are thanked for donation of starting materials and Dr Ivan Prokes is thanked for assistance with NMR spectroscopic measurements.Notes and references
- D. Bourissou, S. Moebs-Sanchez and B. Martin-Vaca, C. R. Chim., 2007, 10, 775–794 CrossRef CAS
.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS
- J. M. Becker, R. J. Pounder and A. P. Dove, Macromol. Rapid Commun., 2010, 31, 1923–1937 CrossRef CAS
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 Search PubMed
- O. Coulembier, P. Degée, J. L. Hedrick and P. Dubois, Prog. Polym. Sci., 2006, 31, 723–747 CrossRef CAS
- G. Rokicki, Prog. Polym. Sci., 2000, 25, 259–342 CrossRef CAS
- S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013 10.1039/c2cs35268k
- E. F. Connor, G. W. Nyce, M. Myers, A. Mock and J. L. Hedrick, J. Am. Chem. Soc., 2002, 124, 914–915 CrossRef CAS
- A. P. Dove, H. B. Li, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2006, 2881–2883 RSC
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, E. C. Hagberg, G. W. Nyce, R. M. Waymouth and J. L. Hedrick, Polymer, 2006, 47, 4018–4025 CrossRef CAS
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS
- M. Fevre, J. Pinaud, A. Leteneur, Y. Gnanou, J. Vignolle, D. Taton, K. Miqueu and J. M. Sotiropoulos, J. Am. Chem. Soc., 2012, 134, 6776–6784 Search PubMed
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798–13799 CrossRef CAS
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. B. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863–7871 CrossRef CAS
- L. Toom, A. Kutt, I. Kaljurand, I. Leito, H. Ottosson, H. Grennberg and A. Gogoll, J. Org. Chem., 2006, 71, 7155–7164 CrossRef CAS
- C. Thomas, F. Peruch, A. Deffieux, A. Milet, J. P. Desvergne and B. Bibal, Adv. Synth. Catal., 2011, 353, 1049–1054 CrossRef CAS
- S. Koeller, J. Kadota, A. Deffieux, F. Peruch, S. Massip, J. M. Leger, J. P. Desvergne and B. Bibal, J.
Am. Chem. Soc., 2009, 131, 15088–15089 CrossRef CAS
- S. Koeller, J. Kadota, F. Peruch, A. Deffieux, N. Pinaud, I. Pianet, S. Massip, J. M. Leger, J. P. Desvergne and B. Bibal, Chem.–Eur. J., 2010, 16, 4196–4205 CrossRef CAS
- A. Alba, A. Schopp, A. P. D. Delgado, R. Cherif-Cheikh, B. Martin-Vaca and D. Bourissou, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 959–965 CrossRef CAS
- O. Coulembier, D. P. Sanders, A. Nelson, A. N. Hollenbeck, H. W. Horn, J. E. Rice, M. Fujiwara, P. Dubois and J. L. Hedrick, Angew. Chem., Int. Ed., 2009, 48, 5170–5173 CrossRef CAS
- T. A. Johnson, M. D. Curtis and P. Beak, Org. Lett., 2002, 4, 2747–2749 CrossRef CAS
- S. D. Wu, S. Lee and P. Beak, J. Am. Chem. Soc., 1996, 118, 715–721 CrossRef CAS
- P. Oña-Burgos, I. Fernández, L. Roces, L. Torre-Fernández, S. García-Granda and F. López-Ortiz, Org. Lett., 2008, 10, 3195–3198 CrossRef CAS
- C. Metallinos and V. Snieckus, Org. Lett., 2002, 4, 1935–1938 CrossRef CAS
-
P. M. Dewick, in Medicinal Natural Products, John Wiley & Sons, Ltd, 2009, pp. 311–420 Search PubMed
- J.-P. R. Hermet, M. J. McGrath, P. O'Brien, D. W. Porter and J. Gilday, Chem. Commun., 2004, 1830–1831 RSC
- N. R. Norcross, J. P. Melbardis, M. F. Solera, M. A. Sephton, C. Kilner, L. N. Zakharov, P. C. Astles, S. L. Warriner and P. R. Blakemore, J. Org. Chem., 2008, 73, 7939–7951 Search PubMed
- D. J. Coady, A. C. Engler, H. W. Horn, K. M. Bajjuri, K. Fukushima, G. O. Jones, A. Nelson, J. E. Rice and J. L. Hedrick, ACS Macro Lett., 2012, 1, 19–22 Search PubMed
- P. C. Ruenitz and E. E. Smissman, J. Heterocycl. Chem., 1976, 13, 1111–1113 Search PubMed
- A. Gogoll, C. Johansson, A. Axén and H. Grennberg, Chem.–Eur. J., 2001, 7, 396–403 CrossRef CAS
- J. A. Landgrebe and L. W. Becker, J. Am. Chem. Soc., 1968, 90, 395–400 Search PubMed
- G. M. Miyake and E. Y. X. Chen, Macromolecules, 2011, 44, 4116–4124 CrossRef CAS
- L. Zhang, F. Nederberg, J. M. Messman, R. C. Pratt, J. L. Hedrick and C. G. Wade, J. Am. Chem. Soc., 2007, 129, 12610–12611 CrossRef CAS
- J. Coudane, C. UstarizPeyret, G. Schwach and M. Vert, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1651–1658 CrossRef CAS
- J. E. Kasperczyk, Polymer, 1999, 40, 5455–5458 CrossRef CAS
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC
- M. T. Zell, B. E. Padden, A. J. Paterick, K. A. M. Thakur, R. T. Kean, M. A. Hillmyer and E. J. Munson, Macromolecules, 2002, 35, 7700–7707 CrossRef CAS
- F. Nederberg, B. G. G. Lohmeijer, F. Leibfarth, R. C. Pratt, J. Choi, A. P. Dove, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2006, 8, 153–160
- S. Tempelaar, L. Mespouille, P. Dubois and A. P. Dove, Macromolecules, 2011, 44, 2084–2091 CrossRef CAS
- D. M. Stevens, S. Tempelaar, A. P. Dove and E. Harth, ACS Macro Lett., 2012, 1, 915–918 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, polymer tacticity analysis, thermal analysis and MALDI-ToF spectra. See DOI: 10.1039/c2sc22053a |
| This journal is © The Royal Society of Chemistry 2013 |