Regio- and stereoretentive synthesis of branched, linear (E)- and (Z)-allyl fluorides from allyl carbonates under Ir-catalysis†
Elena
Benedetto
,
Matthew
Tredwell
,
Charlotte
Hollingworth
,
Tanatorn
Khotavivattana
,
John M.
Brown
* and
Véronique
Gouverneur
*
Chemistry Research Laboratory, University of Oxford, 12, Mansfield Road, OX1 3TA Oxford, UK. E-mail: veronique.gouverneur@chem.ox.ac.uk; john.brown@chem.ox.ac.uk; Fax: +44 (0)1865 285002; Tel: +44 (0)1865 275644
First published on 24th October 2012
Abstract
This paper describes a new catalytic method for the regio- and stereocontrolled fluorination of allylic carbonates. This transformation uses TBAF·4tBuOH as the fluoride source and [Ir(COD)Cl]2 as the catalyst; the most commonly used [Ir(COD)Cl]2/phosphoramidite system is ineffective. Synthetically, this reaction is characterized by a high degree of structural conservation in going from substrates to the products. The fluorination of (E)-allylic carbonates leading to linear (E)-allylic fluorides (l![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :b > 20:1, E:Z > 20:1) is unprecedented and a unique feature of fluoride as the nucleophile. The first examples of transition metal catalyzed fluorination affording (Z)-allyl fluorides (Z:E ratio >20:1) are disclosed along with the successful fluorination of branched, linear (E)- and (Z)-allyl carbonates with [18F] fluoride in the presence of [Ir(COD)Cl]2. 18O-Labeling of the reactant reveals internal return during the allylic ionization step, and pathways for effective intra- and intermolecular isotope exchange.
:b > 20:1, E:Z > 20:1) is unprecedented and a unique feature of fluoride as the nucleophile. The first examples of transition metal catalyzed fluorination affording (Z)-allyl fluorides (Z:E ratio >20:1) are disclosed along with the successful fluorination of branched, linear (E)- and (Z)-allyl carbonates with [18F] fluoride in the presence of [Ir(COD)Cl]2. 18O-Labeling of the reactant reveals internal return during the allylic ionization step, and pathways for effective intra- and intermolecular isotope exchange.
Introduction
Extensive efforts have been dedicated to allylic fluoride synthesis because these versatile building blocks are useful for many chemical disciplines including medicinal chemistry, clinical imaging and material science.1 An ideal toolbox of synthetic methods would allow access to branched and linear allylic fluorides of E or Z geometry with perfect control over regio- and enantioselectivity. Atom economical processes are the most sought after, especially those using readily available starting materials and inexpensive fluoride sources.2 Pursuit of these goals has led to major advances. Our own contribution established that the Pd-catalyzed fluorination of allyl p-nitrobenzoates and carbonates is feasible using TBAF·4tBuOH as the fluoride source. The short fluorination time allowed for the [18F] labeling of allyl carbonate precursors under very mild conditions.3 Doyle and co-workers demonstrated that the combination of allyl chlorides and AgF is highly effective under Pd catalysis for the synthesis of enantioenriched cyclic and acyclic allylic fluorides.4 Cinnamyl phosphorothioate esters are also suitable substrates for regioselective fluorination under Pd catalysis; this reaction is performed in THF and requires AgF as the fluoride source.5 Recently, the unique properties of the trichloroacetimidate leaving group led to the development of the first iridium-catalyzed allylic fluorination leading to terminal branched secondary and tertiary allylic fluorides, the product being formed at the site where the leaving-group departs. Under these conditions, primary allylic trichloracetimidates were unreactive.6 For this reaction, the unligated [Ir(COD)Cl]2 complex (COD = cycloocta-1,5-diene) was used as the catalyst and TEA·3HF was identified as the optimal fluoride source. It was noted that allylic acetates did not react, and allylic carbonates led mainly to undesired allylic ethers. The development of a more general Ir-catalyzed fluorination using readily accessible allylic carbonates is highly desirable because of the distinctive characteristics of Ir versus Pd in allylic substitution, as noted in the initial reports.7 Preferential formation of the more highly branched allylic products from branched and linear E-allylic precursors with C-nucleophiles and heteronucleophiles other than fluoride is well documented for Ir-catalyzed reactions, as is the conservation of regio- and stereochemical integrity for Z-allylic substrates.8,9 Herein, we report that allylic carbonates are amenable to fluorination through iridium catalysis using our originally proposed fluoride source, TBAF·4tBuOH.3 This catalytic process allows access to both branched allylic fluorides as well as their linear E- and Z-regioisomers. In addition to expanding the synthetic value of transition metal catalyzed allylic fluorination with the first examples of (Z)-allyl fluorides, the present body of work reveals unexpected results. The catalytic system [Ir(COD)Cl]2/phosphoramidite,8–10 commonly used in allylic alkylation and amination since the initial discoveries,10 is not effective for fluorination. In addition, unusual regioselectivity trends specific to the use of fluoride as the nucleophile or additive have been observed (Fig. 1).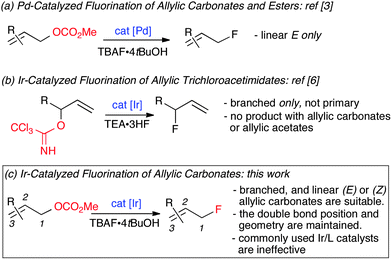 | ||
| Fig. 1 Pd- and Ir-catalyzed allylic fluorination by displacement of O-leaving groups. | ||
Results and discussion
A. Validation and optimization studies
The lack of precedent for Ir-catalyzed substitution of allyl fluorides encouraged us to examine the leaving group propensity of carbonate vs. fluoride in alkylation with malonate. The branched allylic carbonate 1a and branched allylic fluoride 2a were subjected to alkylation with sodium dimethylmalonate in the presence of 2 mol% of [Ir(COD)Cl]2 complex A in THF at 40 °C for 24 h. Both substrates led predominantly to the branched product 3a; branched over linear selectivity was slightly superior for 2a (4:1) in comparison with 1a (2:1). Compound 1a was totally consumed while only 56% of allyl fluoride 2a was converted into products, a pattern indicating that in this system, methyl carbonate is a more reactive leaving group than fluoride. Prior work by our group has shown that a similar order of reactivity was observed under Pd catalysis,11 but fluoride was a more reactive leaving group than methyl carbonate in Pt-catalyzed allylic alkylation (Scheme 1).12
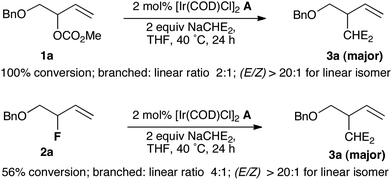 | ||
| Scheme 1 Leaving group propensity of carbonate versus fluoride; NaCHE2 = NaCH(CO2Me)2. | ||
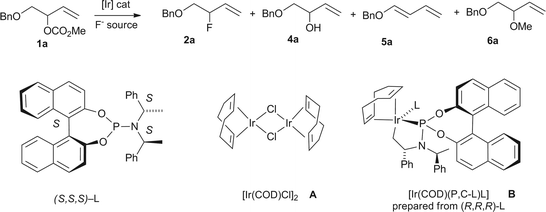
Having established that allyl carbonate 1a is more reactive than allyl fluoride 2a, we embarked on a study aimed at developing a route to 2a from 1a under Ir catalysis. An extensive screening of reaction conditions was instructive and led to the identification of the optimal catalyst, fluoride source and solvent for this transformation (Table 1). In early experiments, the [Ir(COD)Cl]2 complex A was used as the pre-catalyst with two different P-ligands; TBAF·4tBuOH was selected as the fluoride source, based on our previous success in Pd-catalyzed fluorination.3 The use of the phosphoramidite ligand (S,S,S)-L, now established as the norm in Ir catalysis,8–10 was superior to the π-acceptor ligand P(OPh)3, but led, at most, to 25% of the desired allylic fluoride 2a (entries 1 and 2). The use of the pre-formed [Ir(COD)(P,C-L)(L)] catalytic intermediate B prepared from (R,R,R)-L gave a complex reaction mixture, with only trace amount of 2a (entry 3). This was unexpected, and led us to examine the precursor complex [Ir(COD)Cl]2A as a possible source of catalytic turnover. Indeed, this was found to be a much more efficient catalyst (entries 4 and 5). In the absence of Ir catalyst, the starting material was recovered, but contaminated with significant amount of allylic alcohol 4a (∼15%) (entry 6). Some improvement was observed when the reaction was carried out in the presence of molecular sieves (entry 7), but an inferior result was observed at RT (entry 8). The use of TBAF·3H2O afforded the allylic alcohol 4a as the major product (entry 9). HF·pyridine gave a complex product mixture whereas mainly starting material was recovered with CsF (entries 10 and 11). The use of Et3N·3HF led to unreacted starting material and allyl methyl ether, whilst AgF was unreactive (entries 12 and 13). A solvent screen identified DCM as the best solvent for this reaction (entries 14–17). The yield was somewhat improved in the presence of molecular sieves, although more diene 5a was formed.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalystb | F sourcec | Solvent | Time/h | Ratiod1a:2a:4a:5a:6a |
|
a 4 mol% of [Ir], [Ir]:ligand = 1:2; 2 equiv. of fluoride; 1a (0.1 M); 50 °C.
b
A = [Ir(COD)Cl]2, B = [Ir(COD)(P,C-L)(L)], C = [Ir(COD)acac].
c
I = TBAF·4tBuOH, II = TBAF·3H2O, III = HF·pyridine, IV = CsF, V = Et3N·3HF, VI = AgF.
d Analysis by 1H NMR of the crude reaction mixture.
e Addition of 0.1 equiv. of DABCO.
f Complex reaction mixture containing diene 5a.
g Addition of crushed 3 Å sieves.
h At RT.
i Complex reaction mixture.
j At 40 °C.
k At 40 °C, 1a (0.02 M).
l
A added in 4 portions.
|
|||||
| 1 | A/P(OPh)3 | I | THF | 24 | 50:0:25:25:0 |
| 2 | A/(S,S,S)-Le | I | THF | 24 | 38:25:25:12:0 |
| 3 | B | I | THF | 24 | —f |
| 4 | A | I | THF | 5 | 0:55:41:4:0 |
| 5 | C | I | THF | 2 | 7:48:41:4:0 |
| 6 | — | I | THF | 5 | 86:0:14:0:0 |
| 7g | A | I | THF | 5 | 0:69:29:2:0 |
| 8h | A | I | THF | 3 | 33:33:33:1:0 |
| 9h | A | II | THF | 24 | 0:15:81:4:0 |
| 10h | A | III | THF | 24 | —i |
| 11h | A | IV | THF | 24 | 82:0:9:9:0 |
| 12h | A | V | THF | 24 | 63:0:6:1:30 |
| 13h | A | VI | THF | 24 | 100:0:0:0:0 |
| 14 | A | I | DMF | 2 | 24:12:62:2:0 |
| 15 | A | I | Toluene | 2 | 0:61:35:4:0 |
| 16j | A | I | DCM | 2 | 0:80:13:7:0 |
| 17g,j | A | I | DCM | 2 | 0:84:6:10:0 |
| 18k | A | I | DCM | 4 | 18:70:4:8:0 |
| 19k,l | A | I | DCM | 3 | 6:83:5:6:0 |
The allylic alcohol 4a and the diene 5a are consistently seen as side-products but their formation was minimized by conducting the reaction at a higher dilution [1a (0.02 M)] (entry 18). In no instance was the linear allyl fluoride detected in the crude reaction mixture. The appearance of products was monitored by 1H NMR spectroscopy. As the reaction did not progress after 2 h, portion-wise addition of the catalyst (4 portions) was attempted; this protocol did improve the yield of the reaction (entry 19). Further experiments13 showed that alternative leaving groups were less efficient with large recovery of the starting material for allylic benzoate or acetate (>90% starting material, <10% of 2a after 24 h). The fluorination of the allylic p-nitrobenzoate gave 31% of 2a. These initial studies led to the identification of optimal conditions for the reaction: 1a (0.02 M), 2 mol% [Ir(COD)Cl]2A (added in 4 portions), 2 equiv. of TBAF·4tBuOH, DCM, 40 °C, 3 h (as entry 19). Under these conditions, the branched allylic fluoride 2a was isolated in 57% yield.‡
B. Fluorination of linear allylic carbonates (E)- and (Z)-1b
Our next objective was to examine the reactivity of linear allylic carbonates under the preferred reaction conditions (Scheme 2). Fluorination of the isomerically pure linear allylic carbonate (E)-1b afforded the linear allylic fluoride (E)-2b (E:Z > 20:1) in 55% isolated yield (eqn (1)). Careful analysis of the crude reaction mixture indicated that neither allylic methyl ether nor branched product 2a had formed (l:b > 20:1). The high degree of structural conservation observed upon fluorination of (E)-1b is unexpected; indeed, a control experiment demonstrated that (E)-1b gave a mixture of branched and linear product of alkylation 3a and (E)-3b (b:l = 55:45) with sodium dimethylmalonate (eqn (2)). The (Z)-1b isomer also underwent regioselective fluorination affording (Z)-2b in 64% yield, with a high ratio of linear over branched products (l:b > 20:1) and Z selectivity (Z:E > 20:1) (eqn (3)). Similar selectivity was observed for the reaction with sodium dimethylmalonate (eqn (4)). A preliminary investigation suggests that the rate of fluorination followed the order: 1a > (Z)-1b ∼ (E)-1b. Faster reaction rates for branched vs. linear substrates were reported by Helmchen for Ir(I) catalyzed allylic alkylation of allyl acetates with malonate.14
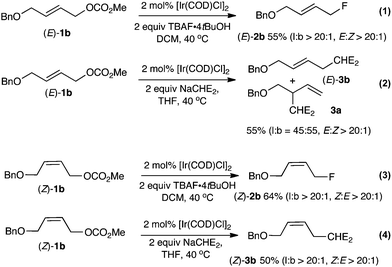 | ||
| Scheme 2 Reactivity of the linear allyl carbonate (E)-1b and (Z)-1b with TBAF·4tBuOH and with NaCHE2 (= NaCH(CO2Me)2). | ||
C. Substrate scope: Ir-catalyzed fluorination of branched and linear allyl carbonates
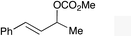
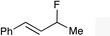
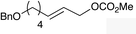
The regioselectivity trend observed upon fluorination of 1a, (E)-1b and (Z)-1b was consistent throughout a series of branched and linear allyl carbonates (Table 2). The branched allyl carbonates 1c–e underwent fluorination with retention of regiochemistry (entries 1–3). The non-terminal branched allylic carbonate (E)-1e gave (E)-4-fluoropent-2-en-1-ylbenzene 2e as a single regio- and E-isomer in 66% yield (NMR) (entry 3). The fluorination of the linear allyl carbonates (E)-1f–h followed the unusual pattern of regiocontrol observed for (E)-1b (entries 4–6). Linear 2f–h were formed exclusively with excellent E-stereoselectivity. Pleasingly, the allylic fluorides (Z)-2i–l were also obtained as pure regio- and Z-isomers from the corresponding allylic carbonates (Z)-1i–l (entries 7–10). The tolerance of these Ir-catalyzed fluorinations is broad since ether, ester, carbamate and imide functional groups can all be accommodated within the substrate.:1) and diastereomer (>20:1)b
| Entry | Allyl carbonate | Product | Yieldc (%) | ||
|---|---|---|---|---|---|
|
a Allyl carbonate (0.02 M), 2 mol% [Ir(COD)Cl]2A, 2 equiv. of TBAF·4tBuOH, DCM, 40 °C, 2–24 h.
b b/l (branched/linear) and E/Z ratio determined by 19F NMR spectroscopy on the crude product.
c Yield of isolated product.
d 4 mol% [Ir(COD)Cl]2A.
e NMR yield.
f l/b ratio = 15:1.
g
E/Z ratio = 1:18.
|
|||||
| 1 |
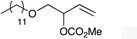
|
1c |
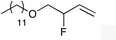
|
2c | 64 |
| 2 |
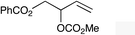
|
1d |
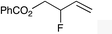
|
2d | 32d |
| 3 |
|
1e |
|
2e | 66d,e |
| 4 |
|
(E)-1f |
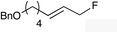
|
(E)-2f | 68 |
| 5 |
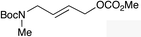
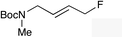
|
(E)-1g |
|
(E)-2g | 64 |
| 6 |
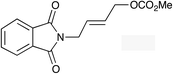
|
(E)-1h |
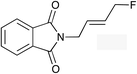
|
(E)-2h | 48f |
| 7 |

|
(Z)-1i |
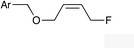
|
(Z)-2i | 52 |
| 8 |
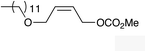
|
(Z)-1j |
|
(Z)-2j | 65 |
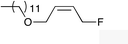
| 9 |
|
(Z)-1k |
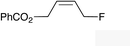
|
(Z)-2k | 65 |
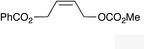
| 10 |
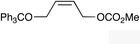
|
(Z)-1l |
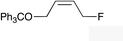
|
(Z)-2l | 50d,g |
For most fluorinations, the major side-product was the allylic alcohol that was separated by silica gel column chromatography.
D. Radiolabeling with [18F] fluoride
The allylic carbonates 1a, (E)-1b and (Z)-1b are amenable to [18F] radiolabeling using either [18F] KF·Kryptofix or [18F] Et4NF[15] as the fluoride source; the results were superior and more reproducible with the later reagent (Scheme 3).13 For the radiofluorination, 1a (5 mg in 440 μl DCM) and [Ir(COD)Cl]2 (60 μl of a 1 mg mL−1 solution in DCM) were added to [18F] Et4NF (∼30 MBq in 30 μl MeCN). After 30 minutes at 40 °C, the reactions were analyzed by radio TLC and HPLC for radiochemical yield (RCY) and product identity, respectively.16 The RCYs were higher for the branched precursor 1a than for the linear precursors; the reaction was found to be significantly more effective for allylic carbonate (E)-1b despite doubling the quantity of [Ir(COD)Cl]2 for (Z)-1b. Control experiments confirmed that no [18F] fluorination took place in the absence of [Ir(COD)Cl]2 for 1a and (E)-1b.![[18F] Fluorination of 1a, (E)-1b and (Z)-1b with [18F] Et4NF in the presence of [Ir(COD)Cl]2.](/image/article/2013/SC/c2sc21789a/c2sc21789a-s3.gif) | ||
| Scheme 3 [18F] Fluorination of 1a, (E)-1b and (Z)-1b with [18F] Et4NF in the presence of [Ir(COD)Cl]2. | ||
E. Probing the origin of regio- and stereoselectivity
The regioselectivity observed for 1a and (E)-1b implies that their reactions did not proceed through similar (η3-allyl) intermediates, or alternatively that the initially formed σ-complex underwent slow σ–π–σ allylic isomerization relative to the rate of fluorination. The sense of regioselectivity observed for the (E)-isomers is unusual under Ir-catalysis, but was documented for Rh-catalyzed17 and Fe-catalyzed18 allylic alkylations. Evans and co-workers provided mechanistic evidence for an enyl organorhodium intermediate accounting for the conservation of regio- (and stereochemistry).17 A σ-allyl intermediate was also advanced for iron-catalyzed allylic alkylation.18 An additional consideration is the influence of the counter-ion on regio- and stereocontrol. Helmchen reported superior b:l selectivity and ee for [Ir(COD)Cl]2 catalyzed allylic alkylation (malonate) of a branched allylic acetate when the reaction was conducted in the presence of 1 equivalent of LiF or LiCl.14 Based on these precedents, we examined the influence of TBAF·4tBuOH as an additive in Ir-catalyzed allylic alkylation of 1a and 1b with malonate (Scheme 4, eqn (1)–(3)). For substrates 1a and (E)-1b, the addition of TBAF·4tBuOH enhanced branched/linear selectivity, favoring a much higher degree of structural conservation. The results are particularly striking for (E)-1b which led exclusively to (E)-3b (l:b ratio > 20:1). We noted that the addition of fluoride had a detrimental effect on the reaction rate as starting material was recovered for all reactions after extended reaction times. A control experiment using tetrabutyl ammonium chloride revealed that this additive had a similar effect on regioselectivity (l:b ratio = 16:1) to TBAF·4tBuOH.13 Mechanistically, we considered the transient formation of an allyl fluoride that would undergo alkylation with malonate; control experiments confirmed that both (E)-2b and (Z)-2b underwent allylic alkylation with the malonate adding to the point of departure of fluoride but the regioselectivity observed for (E)-2b is, however, less pronounced than observed with allyl carbonate/TBAF·4tBuOH (Scheme 4, eqn (4) and (5)).
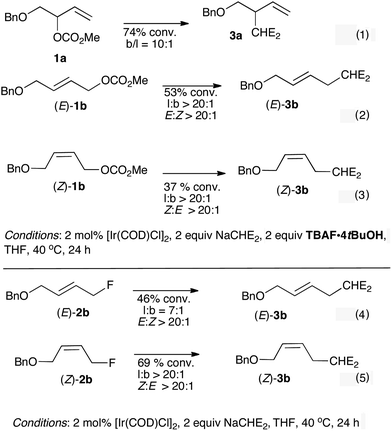 | ||
| Scheme 4 Influence of TBAF·4tBuOH on Ir-catalyzed allylic alkylation of 1a, (E)-1b and (Z)-1b with NaCHE2 (eqn (1)–(3)); reactivity of (E)-2b and (Z)-2b with NaCHE2 under Ir-catalysis (eqn (4) and (5)). | ||
We next examined the fluorination of the branched enantioenriched allylic carbonate (S)-1a (95% ee) (Scheme 5). Under our standard reaction conditions, two products were isolated, the desired allylic fluoride (S)-2a (46% yield, 37% ee) and the allylic alcohol (S)-4a (43% yield, 20% ee). Control experiments conducted in the absence of TBAF·4tBuOH gave unreacted starting material (77%, 87% ee) and diene 5a (23%). When the reaction was conducted at room temperature for 24 h, the allylic fluoride (S)-2a was formed with a higher ee of 48%. Under these milder conditions, the starting material was recovered in 93% ee. Overall retention is the predominant pathway;19 the leakage of stereochemical integrity is significant although far less pronounced than previously observed with trichloroacetimidate as the leaving group.6 The significant degree of racemization is consistent with the formation of a σ-species that could undergo rapid C–C bond rotation prior to fluoride addition. These data contrast with previous studies (using C-nucleophiles) suggesting that the sole use of [Ir(COD)Cl]2 without ancillary ligands typically leads to the highest degree of conservation of enantiomeric purity.14
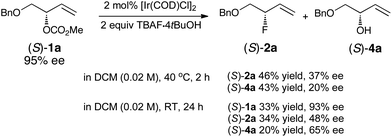 | ||
| Scheme 5 Fluorination of (S)-1a. | ||
F. [18O] Labeling experiments
With the initial intention of understanding the origin of the alcohol side-product, the 18O-isotopomer of allylic methylcarbonate 1b (E:Z 81:19, 98% 18O at C1) was synthesized via the doubly-labeled acetate and alcohol intermediates according to literature guidelines.13,20 Four reactions were conducted with initial analysis aided by the upfield 18O isotopic shift in the quantitative 13C spectrum (CH2O signal):
(i) With labeled 1b under Ir-catalyzed fluorination conditions in DCM, fluoride product 2b was accompanied by recovered but isotope-depleted 1b (33% 18O at C1, similar E:Z ratio), and alcohol 4b (34% 18O at C1).
(ii) Ir-catalyzed fluorination of unlabeled (Z)-1b under standard conditions with 2 equiv. added 18OH2 led to comparable incorporation of label in both recovered reactant and alcohol 4b (ca. 40% 18O at C1).
(iii) The 18O label in enriched 1b (98% 18O at C1) remained intact in the presence of the Ir catalyst under standard reaction conditions, but in the absence of TBAF·4tBuOH.
(iv) Reaction of unlabeled (Z)-1b with TBAF·4tBuOH in the presence of 18OH2 (2 equiv.) at 40 °C overnight led to some hydrolysis (11% (Z)-4b recovered) with no significant label incorporation in either reactant or product.
These results are unexpected, the more so on closer analysis of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O region of the 13C spectrum in the two catalytic turnover experiments (i) and (ii). All four possible oxygen isotopomers of the CH2O–CO component are observed, including significant amounts of the doubly labeled one, confirmed by ES-MS (Fig. 2 and ESI†).21
O region of the 13C spectrum in the two catalytic turnover experiments (i) and (ii). All four possible oxygen isotopomers of the CH2O–CO component are observed, including significant amounts of the doubly labeled one, confirmed by ES-MS (Fig. 2 and ESI†).21
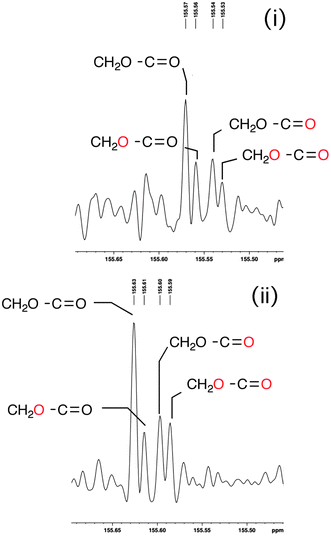 | ||
| Fig. 2 The 13C NMR carbonyl region of recovered (E)-1b following experiment (i) described above and showing the four possible isotopomers; likewise for (Z)-1b in experiment (ii). | ||
This observation minimally requires that an ion-paired intermediate scrambles the label by internal return,22 as has been described for 18O-labeled sulfonates and carboxylates in SN1 reactions. Taken together with the high degree of regio- and stereospecificity observed in allyl fluoride formation, this labeling result supports the reversible intermediacy of an unsymmetrical enyl rather than a fully developed η3-allyl, as has been suggested for Rh-catalyzed allylic substitution.17 In addition, the formation of doubly-labeled and label-depleted recovered reactant in both (i) and (ii) requires a further exchange process between water and the substrate, or between water and a reversibly-formed intermediate. In (i) adventitiously present water is the reagent. The presence of both iridium catalyst and fluoride ion is required for the exchange processes to occur (Scheme 6). The more deep-seated equilibration of 18O labels with external water most likely involves reversible Ir-promoted carbonyl addition of water to the CO double bond. As an analogy for (II), Lloyd-Jones et al. have demonstrated Pd-catalyzed label equilibration between 13CO2 and diallyl carbonate that first involves ionic C–O dissociation.23 The metal-mediated pathway involved in Mosey and Brown's dramatic Pd-catalysed thioamide methanolysis24 offers support for iridium involvement in (I), the intermolecular component of isotope exchange observed here.
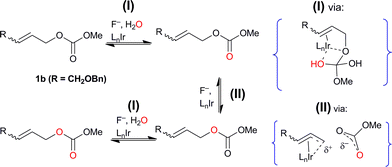 | ||
| Scheme 6 Inter-and intramolecular isotopic exchange observed through 18O-labeling; experiment (ii) with (Z)-1b and 18OH2 used to illustrate likely mechanisms. | ||
G. Synthesis and reactivity of [(allyl)Ir(COD)(P,C-L)(L)] D
Mechanistically, the inability of [Ir(COD)(P,C-L)(L)] B (prepared from (R,R,R)-L) to induce allylic fluorination was unexpected and led us to investigate the origin of this result. Helmchen and co-workers had initially indicated the efficacy of phosphoramidite ligands in Ir allylic alkylation,10a further developed by Hartwig and co-workers10b in their original demonstration of Ir-catalyzed asymmetric allylic aminations. Improvements in the ligand emerged from the Alexakis group.25 Mechanistic insights were contributed by both Helmchen and Hartwig, a key feature being the identification of the isolable reactive intermediate [Ir(COD)(P,C-L)(L)] B formed by a stereospecific internal methyl C–H activation of the phosphoramidite side-chain.26 The cycloocta-1,5-diene (COD) ligand is maintained throughout, allylic carbonates are the preferred reactants, and the stereochemical course follows the palladium case with double inversion leading to overall retention. Based on these solid precedents, the novel allyl Ir-intermediate D was prepared from 1a applying a literature protocol,27 and subjected to three reactions (Scheme 7). Upon treatment of complex D with sodium dimethylmalonate in d8-THF at room temperature followed by addition of PPh3, the branched and linear products of substitution 3a and (E)-3b were obtained in a ∼4:1 ratio, a result corroborating the product distribution observed in the catalytic mode. In contrast, the diene 5a was the major product when exposing complex D to TBAF·4tBuOH, with no signals characteristic of branched allylic fluoride 2a, or Ir–F or P–F bond formation. Under these conditions, complex D had decomposed. The combined use of malonate and TBAF·4tBuOH also led to decomposition of D with 3a (7%), 5a (23%) and a trace amount of (E)-3b (∼3%) detectable in the crude reaction mixture. Collectively, these data demonstrate that the allyl Ir-intermediate D is inactivated by fluoride in d8-THF. Further NMR studies13 indicate that D did not react with TBAF·4tBuOH in d2-DCM at room temperature but decomposed with diene formation after 30 minutes at 40 °C. These results discouraged further work with phosphine-Ir catalysts and encouraged us to focus on [Ir(COD)Cl]2A.
![Synthesis and reactivity of [Ir(COD)(P,C-L)(L)] D.](/image/article/2013/SC/c2sc21789a/c2sc21789a-s7.gif) | ||
| Scheme 7 Synthesis and reactivity of [Ir(COD)(P,C-L)(L)] D. | ||
Conclusions
In conclusion, we have developed a method that converts allyl carbonates into allyl fluorides under iridium catalysis using TBAF·4tBuOH as the fluoride source. This study has unveiled a series of novel features in Ir catalysis and its application to allylic fluorination. The inability of the [Ir(COD)Cl]2/(S,S,S)-L catalytic system to induce fluorination was unexpected; control experiments demonstrated that [Ir(COD)(P,C-L)(L)] is inactivated in the presence of fluoride. This complication was addressed by using the unligated [Ir(COD)Cl]2. The sense and level of regiocontrol for the fluorination of (E)-allyl carbonate leading to linear (E)-allyl fluoride is a distinct characteristic of fluoride as the incoming nucleophile and is unprecedented. This fluoride effect has been extended to allylic alkylation of malonate that delivered linear (E) isomers from linear precursors when the reaction was performed in the presence of TBAF·4tBuOH. The first examples of transition metal catalyzed fluorination affording (Z)-allyl fluorides are disclosed and the [18F] radiolabeling of representative branched and linear (E)- and (Z)-allyl carbonates was successfully validated. Future work aims at identifying the active catalytic Ir-species and our next synthetic challenge is the development of a method using allylic alcohols as starting materials.Acknowledgements
This research was financially supported by the European Union FP7-ITN-238434 (E.B.), the EPSRC (M.T., C.H.), CRUK (M.T.), the Nuffield Foundation (T. K.) and the Royal Thai Government (T. K.). We thank Dr T. D. W. Claridge, Dr B. Odell, Dr J. Wickens, Dr J. S. O. McCullagh (Chemistry Research Laboratory, University of Oxford) for key NMR spectroscopy and mass spectrometry experiments.Notes and references
- (a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS; P. Jeschke, ChemBioChem, 2004, 5, 570–589 Search PubMed; (d) T. Furuya, C. A. Kuttruff and T. Ritter, Curr. Opin. Drug Discovery Dev., 2008, 11, 803–819 CAS; (e) J. M. Brown and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 8610–8614 CrossRef CAS.
- C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929–2942 RSC.
- C. Hollingworth, A. Hazari, M. N. Hopkinson, M. Tredwell, E. Benedetto, M. Huiban, A. D. Gee, J. M. Brown and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 2613–2617 CrossRef CAS.
- (a) M. H. Katcher and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 17402–17404 CrossRef CAS; (b) M. H. Katcher, A. Sha and A. G. Doyle, J. Am. Chem. Soc., 2011, 133, 15902–15905 CrossRef CAS.
- A. M. Lauer and J. Wu, Org. Lett., 2012, 14, 5138–5141 CrossRef CAS.
- J. J. Topczewski, T. J. Tewson and H. M. Nguyen, J. Am. Chem. Soc., 2011, 133, 19318–19321 CrossRef CAS.
- (a) R. Takeuchi and M. Kashio, Angew. Chem., Int. Ed. Engl., 1997, 36, 263–265 CrossRef CAS; (b) R. Takeuchi and M. Kashio, J. Am. Chem. Soc., 1998, 120, 8647–8655 CrossRef CAS.
- For reviews on Ir catalysis of allylic substitution, see: (a) R. Takeuchi, Synlett, 2002, 1954–1965 CrossRef CAS; (b) H. Miyabe and Y. Takemoto, Synlett, 2005, 1641–1655 CAS; (c) R. Takeuchi and S. Kezuka, Synthesis, 2006, 3349–3366 CrossRef CAS; (d) G. Helmchen, A. Dahnz, P. Duebon, M. Schelwies and R. Weihofen, Chem. Commun., 2007, 675–691 RSC; (e) G. Helmchen, Iridium in Organic Synthesis, ed. L. A. Oro and C. Claver, Wiley-VCH, New York, 2009, pp. 211–250 Search PubMed; (f) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef CAS; (g) G. Helmchen, U. Kazmaier and S. Foerster, Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 3rd edn, 2010, pp. 497–641 Search PubMed; (h) J. F. Hartwig and M. J. Pouy, Top. Organomet. Chem., 2011, 34, 169–208 CrossRef CAS; (i) W. B. Liu, J. B. Xia and S. L. You, Top. Organomet. Chem., 2012, 38, 155–207 CrossRef; (j) P. Tosatti, A. Nelson and S. P. Marsden, Org. Biomol. Chem., 2012, 10, 3147–3163 RSC.
- Selected references for heteroatom nucleophiles in Ir-catalyzed allylations: N-nucleophiles: (a) R. Takeuchi, N. Ue, K. Tanabe, K. Yamashita and N. Shiga, J. Am. Chem. Soc., 2001, 123, 9525–9534 CrossRef CAS; (b) R. Weihofen, O. Tverskoy and G. Helmchen, Angew. Chem., Int. Ed., 2006, 45, 5546–5549 CrossRef CAS; (c) Y. Yamashita, A. Gopalarathnam and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 7508–7509 CrossRef CAS; (d) M. Lafrance, M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3470–3473 CrossRef CAS ; O-nucleophiles: (a) C. Shu and J. F. Hartwig, Angew. Chem., Int. Ed., 2004, 43, 4794–4797 Search PubMed ; (b) I. Lyothier, C. Defieber and E. M. Carreira, Angew. Chem., Int. Ed., 2006, 45, 6204–6207 Search PubMed ; (c) M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 5568–5571 Search PubMed ; (d) M. Gartner, S. Mader, K. Seehafer and G. Helmchen, J. Am. Chem. Soc., 2011, 133, 2072–2075 Search PubMed ; S-nucleophiles: (a) Q. L. Xu, W. B. Liu, L. X. Dai and S. L. You, J. Org. Chem., 2010, 75, 4615–4618 Search PubMed ; (b) W. Q. Huang, S. C. Zheng, J. L. Tang and X. M. Zhao, Org. Biomol. Chem., 2011, 9, 7897–7903 Search PubMed.
- For early usage of phosphoramidites in Ir-catalyzed allylic substitutions, see: (a) B. Bartels and G. Helmchen, Chem. Commun., 1999, 741–742 RSC; (b) T. Ohmura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 15164–15165 CrossRef CAS.
- A. Hazari, V. Gouverneur and J. M. Brown, Angew. Chem., Int. Ed., 2009, 48, 1296–1299 CrossRef CAS.
- E. Benedetto, M. Keita, M. Tredwell, C. Hollingworth, J. M. Brown and V. Gouverneur, Organometallics, 2012, 31, 1408–1416 CrossRef CAS.
- For details, see the ESI†.
- B. Bartels, C. Garcia-Yebra, F. Rominger and G. Helmchen, Eur. J. Inorg. Chem., 2002, 2569–2586 CrossRef CAS.
- C. D. Reed, G. G. Launay and M. A. Carroll, J. Fluorine Chem., 2012, 143, 231–237 CrossRef CAS.
- HPLC analysis confirmed that the retention time of the [18F] labeled product corresponds to the cold unlabeled material. The retention times of 2a, (E)-2b and (Z)-2b are too similar (6.9 min, 6.8 min and 6.4 min respectively) to differentiate unambiguously between the three possible [18F] labeled allyl fluoride products. Branched versus linear and E/Z selectivity for the radiolabeling experiments was therefore assigned by analogy with the fluorinations performed using the unlabeled reagent TBAF·4tBuOH.
- P. A. Evans and J. D. Nelson, J. Am. Chem. Soc., 1998, 120, 5581–5582 CrossRef CAS.
- For selected examples, see: (a) B. Plietker, Angew. Chem., Int. Ed., 2006, 45, 1469–1473 CrossRef CAS; (b) B. Plietker, Angew. Chem., Int. Ed., 2006, 45, 6053–6056 CrossRef CAS.
- The absolute configuration was assigned by preparing the allylic fluoride (S)-2a independently using a literature protocol (ref. 4b).
- G. C. Lloyd-Jones and S. Purser, Chem.–Eur. J., 2011, 17, 4724–4726 CrossRef CAS.
- On the basis of literature reports, the expected upfield 13C chemical isotope shift is ca. 0.01 ppm for 18O–CO and ca. 0.03 ppm for O–CO18 in 1b; c.f.
(a) J. R. Everett, Org. Magn. Reson., 1982, 19, 86–88 CrossRef CAS;
(b) J. M. Risley, Magn. Reson. Chem., 1991, 29, 143–147 CrossRef CAS.
- (a) H. L. Goering and J. F. Levy, J. Am. Chem. Soc., 1962, 84, 3853–3857 CrossRef CAS; (b) A. F. Diaz, I. Lazdins and S. Winstein, J. Am. Chem. Soc., 1968, 90, 1904–1905 CrossRef CAS; (c) Y. Tsuji, K. Yatsugi, M. Fujio and Y. Tsuno, Tetrahedron Lett., 1995, 36, 1461–1464 CrossRef CAS; (d) Y. Tsuji, M. M. Toteva, T. L. Amyes and J. P. Richard, Org. Lett., 2004, 6, 3633–3636 CrossRef CAS; (e) Y. Tsuji and J. P. Richard, J. Am. Chem. Soc., 2006, 128, 17139–17145 CrossRef CAS.
- A. Gordillo and G. C. Lloyd-Jones, Chem.–Eur. J., 2012, 18, 2660–2665 CrossRef CAS.
- C. T. Liu, C. I. Maxwell, S. G. Pipe, A. A. Neverov, N. J. Mosey and R. S. Brown, J. Am. Chem. Soc., 2011, 133, 20068–20071 CrossRef CAS.
- (a) K. Tissot-Croset, D. Polet and A. Alexakis, Angew. Chem., Int. Ed., 2004, 43, 2426–2428 CrossRef CAS; (b) D. Polet, A. Alexakis, K. Tissot-Croset, C. Corminboeuf and K. Ditrich, Chem.–Eur. J., 2006, 12, 3596–3609 CrossRef CAS.
- (a) B. Bartels, C. Garcia-Yebra, F. Rominger and G. Helmchen, Eur. J. Inorg. Chem., 2002, 2569–2586 CrossRef CAS; (b) C. A. Kiener, C. T. Shu, C. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 14272–14273 CrossRef CAS; (c) C. T. Shu, A. Leitner and J. F. Hartwig, Angew. Chem., Int. Ed., 2004, 43, 4797–4800 CrossRef CAS; (d) C. Garcia-Yebra, J. P. Janssen, F. Rominger and G. Helmchen, Organometallics, 2004, 23, 5459–5470 CrossRef CAS; (e) G. Lipowsky, N. Miller and G. Helmchen, Angew. Chem., Int. Ed., 2004, 43, 4595–4597 CrossRef CAS; (f) A. Leitner, S. Shekhar, M. J. Pouy and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 15506–15514 CrossRef CAS; (g) D. Markovic and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 11680–11681 CrossRef CAS; (h) S. Spiess, J. A. Raskatov, C. Gnamm, K. Broedner and G. Helmchen, Chem.–Eur. J., 2009, 15, 11087–11090 CrossRef CAS; (i) S. T. Madrahimov, D. Markovic and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 7228–7229 CrossRef CAS; (j) S. T. Madrahimov and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 8136–8147 CrossRef CAS; (k) J. A. Raskatov, M. Jäkel, B. F. Straub, F. Rominger and G. Helmchen, Chem.–Eur. J., 2012, 18, 14314–14328 CrossRef CAS.
- J. A. Raskatov, S. Spiess, C. Gnamm, K. Broedner, F. Rominger and G. Helmchen, Chem.–Eur. J., 2010, 16, 6601–6615 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2sc21789a |
| ‡ During these initial studies, the paper from Nguyen's group that describes an effective catalytic synthesis of branched allylic fluorides from branched allylic trichloracetimidates appeared (ref. 6). |
| This journal is © The Royal Society of Chemistry 2013 |