Studies of iron-mediated Pauson–Khand reactions of 1,1-disubstituted-allenylsilanes: mechanistic implications for a reactive three-membered iron metallacycle†
David R.
Williams
*,
Akshay A.
Shah
,
Shivnath
Mazumder
and
Mu-Hyun
Baik
*
Department of Chemistry, Indiana University, 800 E. Kirkwood Avenue, Bloomington, IN 47405, USA. E-mail: williamd@indiana.edu; mbaik@indiana.edu
First published on 15th October 2012
Abstract
Pauson–Khand reactions of 1,1-disubstituted allenylsilanes have been explored using diiron nonacarbonyl. These studies describe the characterization of three-membered iron metallacycles as reaction-competent intermediates leading to stereoselective formation of highly functionalized 4-alkylidene-2-cyclopenten-1-ones. Studies of the reaction profile provide insights leading to the proposal of a stereoselective mechanistic pathway, which has not been previously explored. Furthermore, these reactions have been examined using computational chemistry, and these efforts have detailed the unique behavior of Fe(CO)4 and its role in Pauson–Khand processes.
Introduction
Our recent studies have explored the development of bifunctional reagents which permit the sequential execution of cross-coupling processes in tandem with other high value reactions.1 A goal of these efforts is the design of versatile methodology which provides for the rapid assembly of molecular complexity. In this regard, the Pauson–Khand reaction (PKR), a formal [2 + 2 + 1] cycloaddition, has emerged as an important development for the synthesis of cyclopentenones via a carbonyldicobalt-mediated carbocyclization of an alkene, an alkyne, and carbon monoxide.2 Intramolecular variants of the PKR are especially effective for the synthesis of polycyclic cyclopentenones and natural products.3Although the initial studies of intermolecular PKR processes using aliphatic and alicyclic olefins generally provided poor results, significant contributions by Cazes,4 Brummond,5 Narasaka,6 and others7 have improved the scope of the reaction including the use of other transition-metal catalysts. Synthetic utility has been substantially enhanced by the use of more reactive allenes in place of alkenes, although issues of regioselectivity can arise based on steric effects imposed by the allenyl substitution pattern.8
In this article, we describe a PKR process which proceeds at ambient temperature using 1,1-disubstituted allenylsilanes and diiron nonacarbonyl. Reactions with a variety of terminal alkynes lead to stereoselective formation of highly functionalized (E)-4-alkylidene-2-cyclopentenones. We have investigated the potential for catalytic turnover of the iron-promoted PKR under elevated CO concentrations. We have undertaken kinetic studies to provide supporting evidence that this variant of the PKR occurs via an alternative mechanistic pathway which does not involve initial complexation of the alkyne component. Our study details the first report of a reaction-competent, three-membered iron metallacycle intermediate which has important implications for understanding of these [2 + 2 + 1] cyclizations, and the stereoselectivity of these iron-mediated processes.
Results and discussion
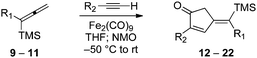
Our initial discovery of regioselective sp2–sp2 Stille cross-coupling reactions of 3-tri-n-butylstannyl-1-trimethylsilyl-1-propyne demonstrate isomerization following the transmetalation of the propargylic stannane 2 (Scheme 1), which results in an allenylpalladium species for reductive coupling to yield 1,1-disubstituted allenylsilanes 3.9 This novel cross-coupling has supplied direct access to a series of conjugated allenes, and we have sought to explore these results in tandem with a mild [2 + 2 + 1] carbocyclization as an efficient route to complex cyclopentenones. Inspired by efforts for target-oriented total synthesis, our initial studies serve to illustrate the two-stage process. Thus, butyrolactone 6 (Fig. 1) is reacted with 2 in the presence of 20 mol% Pd2(dba)3 in dimethylformamide at 22 °C with triphenylarsine (0.8 mmol) and copper(I) iodide (0.8 mmol) additives. Small quantities of lithium chloride (20 mol%) are added to bring the reaction to completion providing a 60% yield of conjugated allene. Subsequent cycloaddition with trimethylsilylacetylene using Fe2(CO)9 at 22 °C in anhydrous THF containing N-methylmorpholine-N-oxide (3.0 equiv.) results in the stereoselective formation of the cyclopentenone 7 (62%). Similarly, the sequence has been employed for direct access to cyclopentenone 8via a sterically demanding [2 + 2 + 1] carbocyclization at room temperature (50% yield; 75% brsm). Based on these early results, we initiated a survey of the scope and generality of the [2 + 2 + 1] cycloaddition using three examples of our 1,1-disubstituted allenylsilanes, 9, 10 and 11, (Fig. 2) with a variety of terminal alkynes. Our results are compiled in Table 1, providing yields of cyclopentenones 12–22 ranging from 50–80% based on starting allene. We note that Narasaka first described the intermolecular Pauson–Khand reactions of allenylsilanes using Fe(CO)4(NMe3) under conditions of photoirradiation.6 Excitation of the metal facilitates the loss of a ligand to open a site for π-coordination of the alkyne or allenylsilane substrate. However, these reactions yielded a mixture of four isomeric 4-alkylidene-2-cyclopenten-1-ones suggesting the characteristic involvement of radical mechanisms in E/Z-alkene equilibrations. Eaton and coworkers have also demonstrated the Fe2(CO)9 mediated [4 + 1] cycloadditions of allenyl imines, allenyl ketones, and diallenes to produce the corresponding five-membered carbonyl derivatives.10 It is proposed that these cycloaddition events are initiated by precoordination of allene as an η2-complex via formation of Fe(CO)4. While other metal carbonyl species, including Co2(CO)8, Mo(CO)6 and W(CO)6 have been reported for [2 + 2 + 1] cycloadditions of allenes, we have observed no reactions at room temperature for 9, 10, and 11 with the usual cobalt and molybdenum reagents, and the use of tungsten hexacarbonyl led to poor conversions (5–10%). The use of solid diiron nonacarbonyl is operationally preferred, however, similar results are achieved with Fe(CO)5 (liquid at 22 °C). Cycloadditions are conducted in anhydrous THF under nitrogen atmosphere and may lead to a reactive Fe(CO)4·THF complex via a disproportionation and/or CO dissociation pathway.11 The use of a CO atmosphere greatly impeded the rate of our reactions, and in some cases, led to no product formation. Rate enhancements are observed by the addition of N-methylmorpholine-N-oxide (NMO) or trimethylamine-N-oxide (TMANO).12 It is anticipated that amine oxides promote a rate acceleration by initial reaction with Fe(CO)5 leading to a vacant coordination site with release of carbon dioxide and amine. Product formation is not observed in dry DMSO, a highly coordinating solvent, whereas our reactions proceed in toluene, albeit at a slower rate.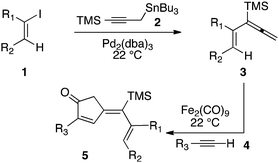 | ||
| Scheme 1 Sequential Stille and Pauson–Khand reactions. | ||
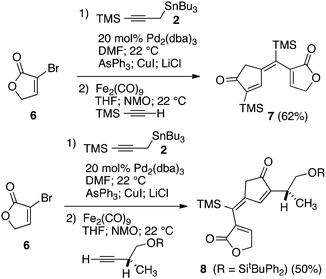 | ||
| Fig. 1 Initial examples of sequential Stille and Pauson–Khand reactions. | ||
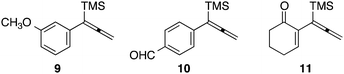 | ||
| Fig. 2 Allenylsilanes 9–11. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Allene | Alkyne | Major producta | Yieldb [isomer/ratio]c |
| a Conditions: to a 0.2 M solution of dry THF containing NMO (3.0 equiv.) was added Fe2(CO)9 (1.5 equiv.) and allene (1.0 equiv.) at −50 °C under N2. Alkyne (1.5–3.0 equiv.) was immediately added with stirring for 5 min, and the mixture was then warmed to rt with stirring for an additional 1–3 h. Dilution with hexanes and filtration through a plug of Florisil® was followed by concentration under reduced pressure to give the crude product. b Yields are quoted for major products after purification by flash silica gel chromatography. c Isomer ratios are based on GC-MS analyses of crude products except for entries 6 and 8, where integrations of selected 1H NMR signals were used. | ||||
| 1 | 9 |

|

|
73% [>97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3] :3] |
| 2 | 9 |

|
|
69% [92:8] |
| 3 | 9 |
|

|
77% [93:7] |
| 4 | 10 |

|

|
81% [94:6] |
| 5 | 10 |
|
|
51% [100:0] |
| 6 | 10 |

|

|
52% [80:20] |
| 7 | 10 |

|

|
61% [>97:3] |
| 8 | 11 |

|
|
74% [83:17] |
| 9 | 11 |
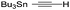
|
|
64% [100:0] |
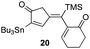
| 10 | 11 |

|
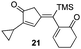
|
72% [>97:3] |
| 11 | 11 |

|

|
61% [97:3] |
In all cases, the products of Table 1 are characterized by the exclusive formation of the exocyclic-(E)-alkenylsilane geometry. The E-stereochemistry of the alkenylsilane has been determined by two-dimensional NOE experiments which established the cis-relationship of TMS and the cyclopentenyl methylene of our products. Furthermore, cyclopentenone 18 (entry 7 of Table 1) afforded suitable crystals leading to an unambiguous structure determination via an X-ray diffraction study.13 Steric effects play an important role in determining the regiochemistry for incorporation of terminal alkynes. As a result, the use of trialkylsilyl and tri-n-butylstannyl acetylenes give rise to the anticipated α-substituted-2-cyclopentenones 12, 15, 16, 18 and 20 (entries 1, 4, 5, 7, 9). On the other hand, reactions with phenylacetylene and alkyl-substituted terminal alkynes (entries 2, 6, 8 and 11; Table 1) generally produced small amounts of the corresponding β-substituted-2-cyclopentenones. These ratios of positional isomers were determined by GC-MS prior to chromatographic purification of the major product. The compatibility of the reaction conditions with numerous functionalities is a particularly valuable feature. The direct incorporation of sensitive vinylsilanes and stannanes, allylic silanes, and halides allows for further site-specific elaborations of the cyclopentenone framework.
Significantly, our studies have provided new insights regarding the reaction mechanism for these Pauson–Khand cycloadditions. Each of the starting allenes 9–11 led to the formation of a product which was first observed at −20 °C and persisted throughout the course of the reaction. In most cases, this substance remained as a minor component of the crude product mixture after consumption of starting allene. Isolation by flash silica gel chromatography and characterization using HRMS and 1H NMR data proved that these isomerically pure substances were adducts of Fe(CO)4 and our starting allenes. This intriguing aspect was investigated by combining 1-(1-napthyl)-1-trimethylsilylallene (23) with diiron nonacarbonyl (1 equiv.) in THF at 22 °C, and led to the isolation of the three-membered iron metallacycle 24 in 41% yield with recovery of starting 23 (55%) (Fig. 3). No other products were obtained. An X-ray diffraction study has afforded an unambiguous structure determination of the complex 24 as illustrated in the ORTEP (Fig. 3).14 In similar fashion, the analogous metallacycle has also been prepared in 67% yield from allene 9.
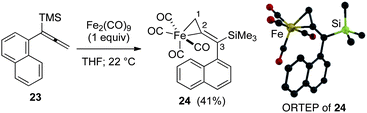 | ||
| Fig. 3 Formation of three-membered metallacycle 24. | ||
Structures from the X-ray determination and density functional theory (DFT) calculations of the three-membered metallacycle have been compared in Table 2. Geometric parameters of the calculations are in good agreement and validate our computational methodology. The results of the X-ray study (ORTEP; Fig. 3) illustrate an expected conformational feature of the complex which places the aryl moiety of the alkenylsilane nearly perpendicular to the plane of the metallacycle. To gain insights with regard to the hybridization at C1 and C2, and to assess the nature of bound iron in the complex, we initially compared the C1–C2 bond length (1.401 Å) of 24 with X-ray crystallographic data for the length of the terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of 1,1-disubstituted allenes (1.29 Å),15 and for the corresponding C1–C2 bond of methylenecyclopropanes (1.467 Å).16 This comparison shows that the C1–C2 bond of 24 is similar to the bonding length which characterizes the analogous carbocycle. In addition, the C1–C2–C3 bond angle (149.21°) of 24 shows considerable deviation from linearity. Thus, an analysis of the X-ray data is in close agreement with our DFT calculations which describe a three-membered metallacycle structure.
C of 1,1-disubstituted allenes (1.29 Å),15 and for the corresponding C1–C2 bond of methylenecyclopropanes (1.467 Å).16 This comparison shows that the C1–C2 bond of 24 is similar to the bonding length which characterizes the analogous carbocycle. In addition, the C1–C2–C3 bond angle (149.21°) of 24 shows considerable deviation from linearity. Thus, an analysis of the X-ray data is in close agreement with our DFT calculations which describe a three-membered metallacycle structure.
For each allene 9–11, these iron metallacyclopropanes were shown to be reaction competent. Reconstitution of our Pauson–Khand reactions with the iron complexes in anhydrous THF with alkyne and NMO (1.0 equiv.) at room temperature led to the rapid production of the expected cyclopentenones without additional requirements of a carbon monoxide atmosphere or elevated temperatures. Reactions, monitored over 30 h in the absence of NMO, showed very slow production of cyclopentenone in the presence of the starting iron metallacycles and alkyne. We attribute this background reaction to a photo-induced activation of the iron complex which allows for loss of one CO ligand. In both instances, small amounts of starting allene were detected in solution.17 The catalytic behavior of these reactions was examined by evaluating three reactions of allene 9 in the presence of 20 mol% Fe2(CO)9 (Table 3). In each case, reagents were combined in THF solution at −60 °C under nitrogen, and were allowed to warm to 22 °C. One reaction (A) was transferred to an autoclave and placed under CO atmosphere (50 psi) for 24 hours. A second reaction (B) was initiated at the same time and under identical conditions as reaction A. However, reaction B was quenched when A was fully pressurized with CO in order to measure the background conversion to product 25 which would occur without the presence of a CO atmosphere. Reaction C was maintained under N2 for 24 hours without the addition of carbon monoxide. Percentages of allene 9 and product 25 were measured by 1H NMR integration relative to an internal standard after removal of paramagnetic iron byproducts by filtration through Florisil®. The data in Table 3 show a stoichiometric dependence on Fe2(CO)9, and in fact, demonstrate that the presence of additional CO effectively halts the progress of the reaction (compare A to C).
|
|
||
|---|---|---|
| Experiment | Conditions | Results |
| A | 50 psi CO atm; 24 h | 25 (10%); 9 (90%) |
| B | N2 atm; 10 min | 25 (10%); 9 (90%) |
| C | N2 atm; 24 h | 25 (18%); 9 (82%) |

Moreover, our kinetic studies have examined the reaction coordinate for consumption of allene, and production of metallacycle and PKR product as a function of time. The PKR reaction profile is shown in Fig. 4. The reactants were dissolved in dry THF at −50 °C under N2 atmosphere, and a small amount of 1,3,5-trimethoxybenzene was added as an internal standard. Aliquots were removed periodically and were immediately passed through a pipette plug of Florisil® using diethyl ether to sequester paramagnetic iron species. Each sample was then analyzed by 1H NMR spectroscopy to measure amounts of allene 9, PKR product 25 and the iron metallacycle 26. Data from three experimental iterations were averaged and graphed. The results display a facile conversion of allene 9 into the cyclopentenone 25 within 50 minutes. However, a small amount of iron metallacycle 26 is rapidly produced and is not consumed as the reaction proceeds to completion. We then examined the production of PKR product 25 and metallacycle 26 as a function of alkyne concentration over linear regions of the initial reaction profile (Fig. 5). In this study, seven reaction vials were charged with Fe2(CO)9, (20 mol%) allene 9 (0.037 mmol), and NMO (3 equiv.) in dry THF at −50 °C under N2 atmosphere. An internal standard of 1,3,5-trimethoxybenzene was added, and various amounts of trimethylsilylacetylene (0.011–0.077 mmols) were introduced at −50 °C followed by warming to 22 °C. After stirring for 60 min, each reaction was diluted with a small amount of hexanes and filtered through a pipette plug of Florisil®. The analysis of these samples by 1H NMR spectroscopy is shown in Fig. 5, and illustrates a linear relationship for the conversion of allene 9 into the cyclopentenone 25 with a steady state amount of metallacycle 26 throughout the range of increasing alkyne concentrations.
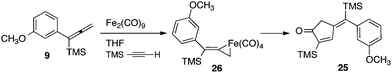
 | ||
| Fig. 4 Reaction profile for the iron-mediated allenic Pauson–Khand reaction (an average of three experimental iterations). | ||
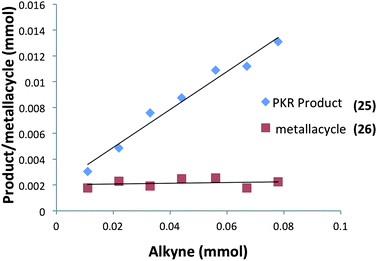 | ||
| Fig. 5 Conversion of allene into PKR product 25 and metallacycle 26 as a function of alkyne concentration (an average of three experimental iterations). | ||
A kinetic study was undertaken for regions of the reaction profile which display linear behavior. Four reaction vessels containing dry THF under N2 atmosphere were charged with NMO (3 equiv.) Fe2(CO)9 (20 mol%), and allene 9 (0.023 mmol) at −50 °C. A quantity of trimethylsilylacetylene (0.014–0.034 mmol) was introduced at −50 °C along with 1,3,5-trimethoxybenzene in THF solution which established a reaction of 0.023 M concentration with respect to allene 9. This concentration ensured reactions were sufficiently slow for periodic removal of aliquots over a timeframe of 10 to 120 minutes. Samples were analyzed by gas chromatography to measure the amount of PKR product 25 leading to the data of Fig. 6.
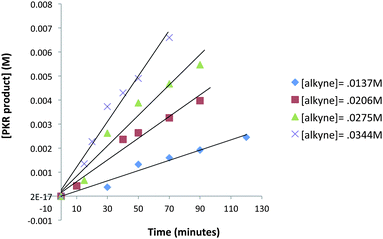 | ||
| Fig. 6 Reaction profile dependence on alkyne concentration in regions which display linear behavior (an average of three experimental iterations). | ||
The rates of each reaction at different alkyne concentrations were determined from the slopes of each plot of Fig. 6. These data are tabulated (Table 4) and display the linear behavior of the graph in Fig. 7. The corresponding log/log plot of data from Table 4 shows that the order of the reaction is 1.6 with respect to alkyne concentration (see ESI†). This behavior is indicative of a complex reaction involving a sequence of steps. In contrast, the kinetic analysis of the classic intermolecular PKR, mediated by CO2(CO)8, demonstrates rate dependence behavior which is zero-order [alkyne].18 Based on these studies, we have concluded that our reactions are stoichiometric with respect to Fe2(CO)9, and suggest that the rate-limiting step is subsequent to the formation of the iron metallacycle. Our reaction profile is not characteristic of the classical PKR and supports evidence of an alternative mechanistic pathway.
| Entry | Alkyne (M) | Initial rate (mmol min−1 mL−1) | Log[alkyne] | Log(rate) |
|---|---|---|---|---|
| 1 | 0.0137 | 2.132 × 10−5 | −1.86 | −4.67 |
| 2 | 0.0206 | 4.517 × 10−5 | −1.69 | −4.35 |
| 3 | 0.0275 | 6.329 × 10−5 | −1.56 | −4.19 |
| 4 | 0.0344 | 9.479 × 10−5 | −1.46 | −4.02 |
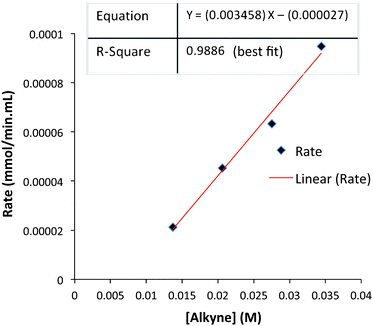 | ||
| Fig. 7 Initial reaction rate as a function of alkyne concentration. | ||
Our findings describe the formation of stable tetracarbonyliron–alkene complexes. There has been long-standing interest in the interactions of allenes with transition-metal reagents, and particularly the ability to distinguish between π-bonding and metallacycle formation.19 It appears that Pettit may have isolated a stable three-membered iron metallacycle from reactions of tetramethylallene and diiron nonacarbonyl as early as 1967. Although this product was not characterized, it was described as a stable π-complex.20 Subsequently, Foxman described the tetramethylallene derivative [Cp(CO)2Fe(η2-Me2CCCMe2)].21 Proton NMR studies showed that the four methyl groups become equivalent with increasing temperature. This behavior suggests rotation about the Fe-allene axis and migration of Cp(CO)2Fe between both orthogonal alkene units. Wojcicki and Lichtenberg reported that η2-allenes derived from coordination of Cp(CO)2Fe undergo nucleophilic attack at the terminal carbon to yield vinylic iron complexes.22 In related studies, Lindner, et al. found that the complexes L(CO)3Fe(η2–C2H4) are obtained by nucleophilic reactions of the bis-triflate of ethylene glycol with the anions [Fe(CO)3L]−2 (where L = PPh3 or POMe3).23 More recently, Sappa and coworkers have described the stable diiron complex Fe2(CO)6(PPh3)[μ-η3-(H2CCCH2)] which has been characterized by X-ray analysis.24 Kukolich and coworkers have reported extensive DFT calculations and low temperature microwave spectroscopy to characterize tetracarbonyl-ethyleneiron,25 and their findings have been compared to structural details stemming from X-ray diffraction data of stable tetracarbonylethyleneosmium, which exhibits evidence of greater back-bonding donation from osmium.26
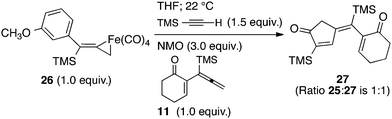
To gain insights into the fundamental reactivity of our iron metallacycles, we designed a crossover experiment in which the purified complex 26 (1 equiv.) was allowed to react with trimethylsilylacetylene (1.5 equiv.) in anhydrous THF at 22 °C with the addition of NMO (3 equiv.) in the presence of the alternative allenylsilane 11 (1 equiv.). Upon completion, this experiment produced approximately equal amounts of two PKR products, the previously characterized 25 and 27 (ratio 1:1). Analyses by 1H NMR spectroscopy and GCMS also showed equal amounts of the starting allenylsilanes 9 and 11 remaining in solution. Our crossover experiment indicates a facile and highly reversible coordination of Fe(CO)4 with the π-system of the allene. This equilibration leads to statistically equal amounts of iron tetracarbonyl complexes derived from 9 and 11 which yield the corresponding cyclopentenones products.
A mechanistic rationale for the PKR was first proposed by Magnus and coworkers in 1985 based upon the precomplexation of Co2(CO)8 with alkyne reagents,27 and this aspect has been broadly applied with additional insights as a general pathway for [2 + 2 + 1] cycloadditions.4a,28 However, our experiments suggest an alternative mechanism resulting from high facial selectivity for the initial complexation leading to the E-alkene geometry of the three-membered metallacycle 28 (Scheme 2). By considering the iron complex 28 as a reactive species for the PKR, we have illustrated two possible pathways leading to cyclopentenone formation. One hypothesis features a stereospecific insertion of carbon monoxide involving bond (b) of 28 which would provide a four-membered intermediate with an open coordination site for alkyne complexation in 29. Subsequent insertion involving bond (a) leads to the acyl metal species 30 for reductive elimination to cyclopentenone product. Alternatively, the loss of carbon monoxide from metallacycle 28 would provide a vacant coordination site for alkyne complexation in 31 followed by selective insertion involving the vinylic bond (a) to produce 32 for conversion to the acyl intermediate 30. In either case, the differential and selective reactions of the allylmetal bond (b) versus the alkenylmetal linkage (a) in 28, in combination with the observed face selectivity for complexation of Fe(CO)4 with the starting allenylsilanes, would suggest new opportunities to explore the chemical behavior of these three-membered metallacycles.
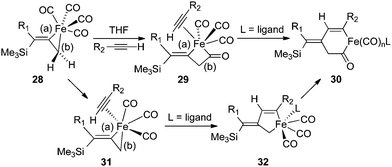 | ||
| Scheme 2 Possible mechanistic pathways involving 28. | ||
To examine these questions and probe the mechanism of the iron-mediated Pauson–Khand reaction, we have employed computational methods based on DFT.29 The dissociation of Fe2(CO)9 is thermodynamically advantageous by approximately 16 kcal mol−1, and leads to two fragments, Fe(CO)4 and Fe(CO)5. The coordinatively unsaturated iron tetracarbonyl species binds the allene, and our calculations suggest that this process is energy neutral. Thus, it is anticipated that Fe(CO)4 complexation with allene substrate is likely to be reversible, and this conclusion is supported by our crossover experiments. The formation of a stable, nearly octahedral, six-coordinate tetracarbonyliron–allene complex poses questions about the nature of the interaction of electrophilic iron species with alkenes. Specifically, these questions address the ability to distinguish between π-complex and ferracyclopropane formation. Our calculations have provided insights which describe the bonding of allene and Fe(CO)4 components. As summarized in Fig. 8, the 1a1 and 2a1 orbitals of Fe(CO)4 have the correct symmetry to overlap with the occupied π-orbital of allene 9. These interactions are viewed as forward donation where electron density is shifted from the CC region toward the metal. Back donation is represented by the overlap of the filled b2 orbital of Fe(CO)4 and the empty π* orbital of the allene. Both forward and back donations play a role in activating the allene for further chemistry.
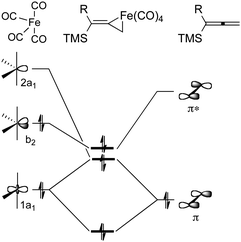 | ||
| Fig. 8 Schematic orbital interaction diagram between Fe(CO)4 and allenylsilane. | ||
DFT calculations reveal five d-type Fe-based molecular orbitals (MO) in the complex 26 (Fig. 9). Molecular orbitals 88 and 89 are regarded as dxz and dyz orbitals, respectively. The mixing of dxy and dx2−y2 gives rise to the MOs 91 and 94. These four MOs are occupied in both α and β subspaces while the higher energy MO 100, which is essentially the dz2 orbital of Fe, is unoccupied. Thus, it is reasonable to classify compound 26 as a d8 Fe° complex. We conclude that bonding of the allenic π-system is so significantly distorted that this arrangement cannot be described as a π-complex. However, it is also not reasonable to describe 26 as a ferracyclopropane which is achieved by the inclusion of a suitably complexed Fe2+ species. The reality lies between these opposing models.
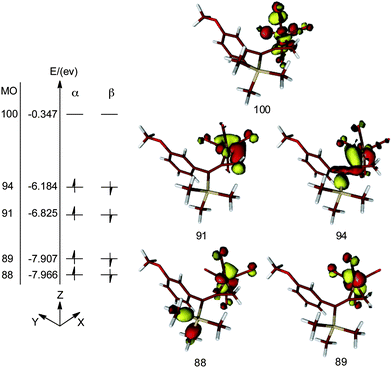 | ||
| Fig. 9 DFT calculated molecular orbitals of the metallacycle 26. Energy states are not drawn to scale. | ||
Our experimental studies have shown that Fe(CO)4 complexation occurs in a regioselective, as well as a stereoselective fashion. DFT calculations have examined the reasons for this behavior. For example, the reaction of Fe2(CO)9 with allene 10 leads to exclusive formation of the complex 33 (Fig. 10). Our calculations show that the more substituted isomer 34 is thermodynamically less stable by 6.5 kcal mol−1. This energy difference may be attributed to two factors. Firstly, the conjugation of the C2–C3 double bond of the starting allene with the aryl substituent may reduce the reactivity of this π-bond for coordination. Secondly, steric repulsions in the ferracyclopropane complex are minimized in 33 as compared to 34.
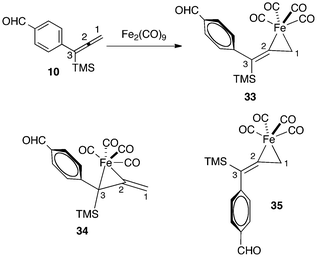 | ||
| Fig. 10 Regio- and stereoselectivity for complexation of allene 10 with Fe(CO)4. | ||
To assess these factors, we have computationally explored the replacement of aryl and trimethylsilyl substituents of 33 and 34 with sterically less demanding vinyl and methyl substitution, respectively (see ESI†). Furthermore, we have calculated the thermodynamic preference for complexation of Fe(CO)4 with 1,1-dimethylallene. In both instances, regioselective coordination is predicted to occur at the terminal olefin.30 We have also examined the face selective outcome which leads to the E-alkene C2–C3 geometry in 33versus the Z-geometry of 35. DFT calculated geometries confirm that the aryl moiety is nearly perpendicular to the plane of the ferracyclopropane in 33, and this conformation avoids steric interactions with neighboring CO ligands. On the other hand, the TMS functionality has a spherical topology which occupies greater spatial volume than aryl. In fact, the replacement of TMS with a methyl group reverses the trend. As shown in Fig. 11, the latter calculations favor isomer 37 in which the optimized geometry permits π-conjugation of the aromatic ring as compared to 36.
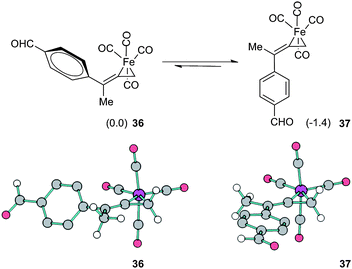 | ||
| Fig. 11 Model study of face selectivity for the initial complexation event. Structures 36 and 37 are also shown with ball and stick illustrations; hydrogen atoms on the aryl ring are omitted for clarity. | ||
Our calculations have provided valuable insights regarding the mechanistic pathways outlined in Scheme 2. Computations show that the insertion of carbon monoxide from 33 (Fig. 12) which leads to a ring expansion to 38, is highly endothermic by 32.7 kcal mol−1. This can be attributed to stable properties of the relatively strain-free and coordinately saturated complex 33. However, the coordination of one THF molecule in 39 is not energetically helpful, and underscores an increase in ring strain associated with this four-membered system compared to the starting 33.
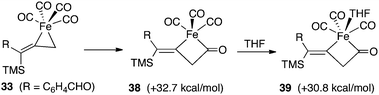 | ||
| Fig. 12 Relative energies leading to ferracyclobutanone 39. | ||
A direct dissociation of CO from 33 provides an open coordination site and results in a sixteen-electron, square pyramidal intermediate. The process is energetically uphill by +20.7 kcal mol−1, and this tight ligand binding explains the absence of catalytic turnover when a CO atmosphere is applied. On the other hand, the introduction of NMO provides for nucleophilic attack on the complex 33 which leads to a fragmentation producing carbon dioxide, N-methylmorpholine and the coordinately unsaturated species 40. This conversion is significantly exothermic by 50.6 kcal mol−1. When we consider filling the coordination sphere to give 41, the overall process is favorable by 51.9 kcal mol−1.
Fig. 13 summarizes our calculated reaction energy profiles for the insertion of alkynes using trimethylsilylacetylene. Although the coordination of THF in 41 (Fig. 14) is downhill by 1.3 kcal mol−1, alkyne binding is slightly endothermic by 1.7 kcal mol−1 to produce π-complexes, such as 42. We have considered four orientations for this complexation leading to the isomeric ferracyclopentenes 43, 44, 45, and 46. The alkyne insertion event is exothermic by 4.6 to 10.3 kcal mol−1, and favored intermediates 43 and 45 present a six-coordinate, 18-electron Fe(II) species with a four electron allenic donor (fragment C1–C2–C3) displaying η3 coordination. In structure 43, the C1–C2 bond distance (1.422 Å) is essentially equal to the C2–C3 distance (1.423 Å). We have examined the transition state arrangements of 47-TS1 and 47-TS3 for insertion of trimethylsilylacetylene into the alkenyl–Fe linkage as well as 47-TS2 and 47-TS4 arrangements for insertion into the allyl–Fe bond as pertains to the orientation and reactivity of the π-complexation shown as 42. Our calculations find that the processes stemming from insertion of the allylmetal linkage via47-TS2 and 47-TS4 require activation energies of 19.5 and 23.7 kcal mol−1 while insertions into the alkenyl–Fe bond to yield 43 and 45 occur with activation barriers of 12.3 and 23.7 kcal mol−1, respectively. The lowest energy pathway involves arrangement 47-TS1 (12.3 kcal mol−1) to produce intermediate 43. Gratifyingly, this regio- and stereoselective insertion gives rise to the experimentally observed cyclopentenones of Table 1. Other insertion pathways are much higher in energy, and thus, imply the greatly preferred formation of α-substituted-2-cyclopentenones.
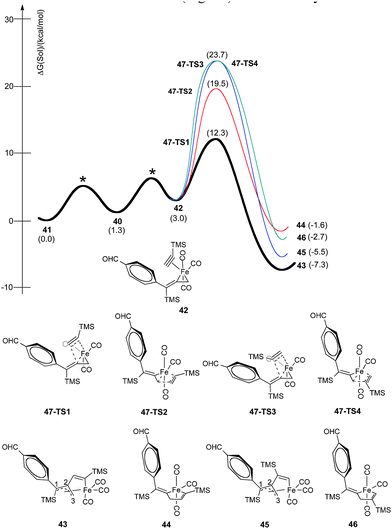 | ||
| Fig. 13 Reaction energy profiles for stereoselective insertion of trimethylsilylacetylene via42 for production of metallacycles 43–46. | ||
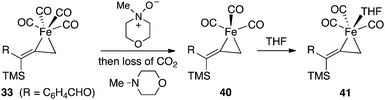 | ||
| Fig. 14 Decarboxylation of 36 with NMO. | ||
To investigate fundamental aspects of this reactivity pattern, we have optimized transition states for the arrangements of 47-TS1 and 47-TS2. As shown in Fig. 15, the transition state stemming from 47-TS2 displays a five-coordinate Fe center as a trigonal bipyramidal structure 49 while 47-TS1 has a classic, three-legged piano stool geometry31 in transition state 48. Bond formation occurs between C2 and C4 in 48, and the calculated Fe–C2 and Fe–C4 bond distances are 2.057 Å and 2.134 Å, respectively. New bond formation must proceed between C3 and C4 atoms in 49 although Fe–C3 and Fe–C4 bond distances are significantly longer at 2.213 Å and 2.237 Å, respectively.
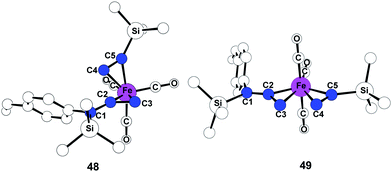 | ||
| Fig. 15 Optimized geometries 48 and 49 of alkyne insertion transition states 47-TS1 and 47-TS2. Hydrogen atoms are omitted for clarity. | ||
In transition state 48, a carbonyl moiety is positioned trans to the bound alkyne, and this feature has important electronic consequences. As a result, the empty dz2 orbital of Fe is positioned to overlap with the filled π orbital of the alkyne as illustrated in 50 (Fig. 16). This attractive interaction effectively lowers the activation energy (12.3 kcal mol−1) of the insertion such that the impending cyclization to metallacycle 43 occurs under very mild conditions. In contrast, structure 49 positions the empty dz2 orbital parallel to the filled π system as shown in 51, and this effect is destabilizing. Further calculations involving the arrangements shown in 47-TS3 and 47-TS4 (Fig. 13) have highlighted destabilizing steric interactions associated with the relative position of the large TMS substituent at the site of C–C bond formation as compared to 47-TS1 and 47-TS2, respectively. Overall, our computational studies have identified a low energy pathway for the iron-mediated PKR process which proceeds via the three-membered d8 Fe° complex 28. As illustrated in Fig. 17, reaction of 28 with NMO provides for decarboxylation and an open coordination site in the reactive complex 40. Addition of alkyne gives 42 which undergoes insertion to 43. Final transformation involves insertion of CO (highlighted in red) to yield complex 52 leading to reductive elimination and formation of the α-substituted-2-cyclopenten-1-one product.
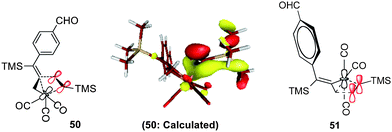 | ||
| Fig. 16 Electronic considerations in 50 compared to 51. | ||
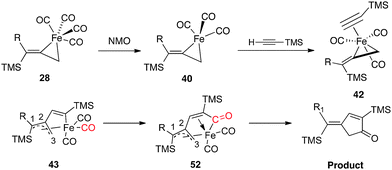 | ||
| Fig. 17 Mechanistic pathway of the iron-mediated PKR. | ||
Conclusions
In summary, a broad-based investigation of iron-mediated [2 + 2 + 1] carbocyclization reactions has been described. An efficient PKR process occurs at ambient temperature using diiron nonacarbonyl, an inexpensive and readily available reagent. In this manner, highly functionalized 4-alkylidene-2-cyclopenten-1-ones are directly available. The process is initiated by a novel Stille cross-coupling of 3-tri-n-butylstannyl-1-trimethylsilyl-1-propyne leading to 1,1-disubstituted allenylsilanes for stereoselective Pauson–Khand reactions. Our studies describe reaction-competent, three-membered metallacycles involving tetracarbonyliron and allene substrate. These metallacycles have electronic properties which characterize d8 Fe° complexes versus π-complexes of alkenes with Fe*2 cation. Our results have shown that increasing concentrations of CO do not support a catalytic turnover, and effectively halt the progress of the reaction. Kinetics display a linear response which is 1.6 order with respect to alkyne concentration. We have detailed a mechanistic pathway describing regioselective, as well as the stereoselective transformations of the metallacycle. Alkyne insertion involves the geometry of a preferred transition state arrangement which features an important stabilizing electronic interaction. Our computational studies conclude that the arrangement of stereochemistry of bound ligands at the transition metal center is crucial for C–C bond formation. These findings have significant ramifications for the design of iron catalysts for cross-coupling processes. Overall, we have documented an alternative pathway demonstrating the role of Fe(CO)4 in novel PKR processes which occur under extraordinarily mild conditions.Acknowledgements
The authors acknowledge the support of the National Science Foundation (CHE-1055441, D.R.W), (CHE-0645381, M.-H.B.) and (CHE-1001589, M.-H.B.).Notes and references
- For examples: (a) D. R. Williams and M. W. Fultz, J. Am. Chem. Soc., 2005, 127(42), 14550–14551 CrossRef CAS; (b) D. R. Williams, A. I. Morales-Ramos and C. M. Williams, Org. Lett., 2006, 8(20), 4393–4396 CrossRef CAS; (c) D. R. Williams and K. G. Meyer, J. Am. Chem. Soc., 2001, 123(4), 765–767 CrossRef CAS.
- (a) I. U. Khand, G. R. Knox, P. L. Pauson, W. E. Watts and M. I. Foreman, J. Chem. Soc., Perkin Trans. 1, 1973, 977–981 RSC; (b) For recent reviews: N. Jeong, Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, UK, 2006, vol. 11, pp. 335–366 Search PubMed; (c) S. Laschat, A. Becheanu, T. Bell and A. Baro, Synlett, 2005, 17, 2547–2570 Search PubMed; (d) S. E. Gibson and N. Mainolfi, Angew. Chem., Int. Ed., 2005, 44(20), 3022–3037 CrossRef CAS.
- For selected examples: (a) Y. Kavanagh, M. O'Brien and P. Evans, Tetrahedron, 2009, 65(39), 8259–8268 Search PubMed; (b) K. A. Miller, C. S. Shanahan and S. F. Martin, Tetrahedron, 2008, 64(29), 6884–6900 CrossRef CAS; (c) K. Kaneda and T. Honda, Tetrahedron, 2008, 64(51), 11589–11593 CrossRef CAS; (d) C. E. Mandu and C. J. Lovely, Org. Lett., 2007, 9(23), 4697–4700 CrossRef CAS; (e) J. Cassayre, F. Gagosz and S. Z. Zard, Angew. Chem., Int. Ed., 2002, 41(10), 1783–1785 CrossRef CAS; (f) J. Chan and T. F. Jamison, J. Am. Chem. Soc., 2004, 126(34), 10682–10691 CrossRef CAS.
- (a) F. Antras, S. Laurent, M. Ahmer, H. Chermette and B. Cazes, Eur. J. Org. Chem., 2010, 3312–3336 Search PubMed; (b) F. Antras, M. Ahmer and B. Cazes, Tetrahedron Lett., 2001, 42(46), 8153–8156 CrossRef CAS; (c) F. Antras, M. Ahmer and B. Cazes, Tetrahedron Lett., 2001, 42(46), 8157–8160 Search PubMed.
- (a) For an overview: K. M. Brummond and J. L. Kent, Tetrahedron, 2000, 56(21), 3263–3283 Search PubMed; (b) K. M. Brummond, H. Chen, K. D. Fisher, A. D. Kerekes, B. Richards, P. C. Still and S. J. Geib, Org. Lett., 2002, 4(11), 1931–1934 CrossRef CAS.
- T. Shibata, Y. Koga and K. Narasaka, Bull. Chem. Soc. Jpn., 1995, 68(3), 911–919 CAS.
- (a) For reviews of rhodium-catalyzed [2 + 2 + 1] carbocyclizations: N. Jeong, Modern Rhodium-Catalyzed Organic Reactions, ed. P. A. Evans, Wiley-VCH, Weinheim, 2005, pp. 215–240 Search PubMed; (b) P. A. Inglesby and P. A. Evans, Chem. Soc. Rev., 2010, 39(8), 2791–2805 RSC.
- For an overview: B. Alcaide and P. Almendros, Eur. J. Org. Chem., 2004, 3377–3383 Search PubMed.
- D. R. Williams and A. A. Shah, Chem. Commun., 2010, 46(24), 4297–4299 RSC.
- (a) M. S. Sigman and B. E. Eaton, J. Org. Chem., 1994, 59(24), 7488–7491 CrossRef CAS; (b) M. S. Sigman, B. E. Eaton, J. D. Heise and C. P. Kubiak, Organometallics, 1996, 15(12), 2829–2832 CrossRef CAS; (c) M. S. Sigman and B. E. Eaton, J. Am. Chem. Soc., 1996, 118(47), 11783–11788 CrossRef CAS.
- F. A. Cotton and J. M. Troup, J. Am. Chem. Soc., 1974, 96(11), 3438–3443 CrossRef CAS.
- (a) J.-K. Shen, Y.-L. Shi, Y.-C. Gao, Q.-Z. Shi and F. Basolo, J. Am. Chem. Soc., 1988, 110(8), 2414–2418 CrossRef CAS; (b) S. Shambayati, W. E. Crowe and S. L. Schreiber, Tetrahedron Lett., 1990, 31(37), 5289–5292 CrossRef CAS; (c) N. Jeong, Y. K. Chung, B. Y. Lee, S. H. Lee and S.-E. Yoo, Synlett, 1991, 3, 204–206 Search PubMed; (d) J.-K. Shen, Y.-C. Gao, Q.-Z. Shi and F. Basolo, Organometallics, 1989, 8(9), 2144–2147 CrossRef.
- The cyclopentenone 18 (C25H38O2Si2) was isolated as pale yellow plates, monoclinic, P1 21/n1, a = 12.129(3) Å, b = 8.192(2) Å, c = 26.318(6) Å, β = 102.587(5)°, V = 2552.2(11) Å3, ρcalc = 1.111 Mg m−3, μ = 0.835 mm−1, 150 K, Mo Kα. A total of 17208 reflections were collected of which 4475 (Rint = 0.075) were unique. Final residues were R1 = 0.0491, wR2 = 0.1064. The structure was solved with direct methods and refined with full-matrix least squares/difference Fourier cycles. All non-hydrogen atoms were refined with anisotropic displacement parameters.
- The iron metallacycle 24 was a liquid sample at room temperature. Upon cooling, colorless crystals were formed and mounted on the tip of a glass capillary at 150 K for X-ray diffraction studies. Crystal data for 24: colorless, acicular crystal, 0.491 × 0.124 × 0.114 mm3, C20H18Fe1O4Si1M = 406.29, monoclinic P21, a = 7.1510(8) Å, b = 29.386(3) Å, c = 9.6851(10) Å, β = 100.773(2)°, V = 1999.4(4) Å3, ρcalc = 1.350 Mg m−3, μ = 0.835 mm−1, 150 K, Mo Kα, 2θmax = 60°. A total of 21632 reflections were collected of which 11113 (Rint = 0.026) were unique. Final residues were R1 = 0.0358, wR2 = 0.0852 (for 9275 observed reflections with I > 2σ(I) and 469 parameters, 1 restraint) with GOF 0.995, and largest difference peak 0.61e Å−3. Data were collected on an APEX II Kappa Duo diffractometer (Bruker). The intensity data were corrected for absorption. Final cell constants were calculated from 9976 strong reflections (APEX II, Bruker-AXS, Madison WI, 2007). The structure was solved with direct methods and refined with full-matrix least squares/difference Fourier cycles. All non-hydrogen atoms were refined with anisotropic displacement parameters.
- (a) Y. Shi, J. Huang, Y.-F. Yang, L.-Y. Wu, Y.-N. Niu, P.-F. Huo, X.-Y. Liu and Y.-M. Liang, Adv. Synth. Catal., 2009, 351(1–2), 141–146 CrossRef CAS; (b) L. F. Tietze, J. R. Wünsch and M. Noltemeyer, Tetrahedron, 1992, 48(11), 2081–2099 Search PubMed.
- A. de Meijere, M. von Seebach, S. I. Kozhushkov, R. Boese, D. Bläser, S. Cicchi, T. Dimoulas and A. Brandi, Eur. J. Org. Chem., 2001, 3789–3795 CrossRef CAS.
- Our NMR studies have not detected the formation of analogous tetracarbonyliron–alkyne complexes in solutions of diiron nonacarbonyl and terminal alkynes.
- R. Cabot, A. Lledó, M. Revés, A. Riera and X. Verdaguer, Organometallics, 2007, 26(5), 1134–1142 CrossRef CAS.
- J. A. Osborn, Chem. Commun., 1968, 1231–1232 RSC.
- R. Ben-Shoshan and R. Pettit, J. Am. Chem. Soc., 1967, 89(9), 2231–2232 CrossRef CAS.
- B. M. Foxman, J. Chem. Soc., Chem. Commun., 1975, 6, 221–222 Search PubMed.
- D. W. Lichtenberg and A. Wojcicki, J. Organomet. Chem., 1975, 94(2), 311–326 CrossRef CAS.
- E. Lindner, E. Schauβ, W. Hiller and R. Fawzi, Chem. Ber., 1985, 118(10), 3915–3931 Search PubMed.
- G. Gervasio, D. Marabello, E. Sappa and A. Secco, Can. J. Chem., 2006, 84(2), 337–344 Search PubMed.
- B. J. Drouin and S. G. Kukolich, J. Am. Chem. Soc., 1999, 121(16), 4023–4030 Search PubMed.
- C. Karunatilaka, B. S. Tackett, J. Washington and S. G. Kukolich, J. Am. Chem. Soc., 2007, 129(34), 10522–10530 Search PubMed.
- (a) P. Magnus and L. M. Principe, Tetrahedron Lett., 1985, 26(40), 4851–4854 CrossRef CAS; (b) P. Magnus, C. Exon and P. Albaught-Robertson, Tetrahedron, 1985, 41(24), 5861–5869 CrossRef CAS.
- (a) V. Derdau, S. Laschat and P. G. Jones, Eur. J. Org. Chem., 2000, 681–689 CrossRef; (b) A. S. Bayden, K. M. Brummond and K. D. Jordon, Organometallics, 2006, 25(22), 5204–5206 CrossRef CAS; (c) For relevant mechanistic discussions of Rh-catalyzed reactions: M.-H. Baik, S. Mazumder, P. Ricci, J. R. Sawyer, Y.-G. Song, H. Wang and P. A. Evans, J. Am. Chem. Soc., 2011, 133(20), 7621–7623 Search PubMed.
- For related DFT calculations: (a) P. Liu and K. N. Houk, Inorg. Chim. Acta, 2011, 369(1), 2–14 CrossRef CAS; (b) M. Yamanaka and E. Nakamura, J. Am. Chem. Soc., 2001, 123(8), 1703–1708 CrossRef CAS; (c) Y. Lan, L. Deng, J. Liu, C. Wang, O. Wiest, Z. Yang and Y.-D. Wu, J. Org. Chem., 2009, 74(14), 5049–5058 CrossRef CAS; (d) C. Wang and Y.-D. Wu, Organometallics, 2008, 27(23), 6152–6162 Search PubMed; (e) X. Verdaguer, J. Vázquez, G. Fuster, V. Bernardes-Génisson, A. E. Greene, A. Moyano, M. A. Pericas and A. Riera, J. Org. Chem., 1998, 63(20), 7037–7052 CrossRef CAS; (f) W. Imhof, E. Anders, A. Göbel and H. Görls, Chem.–Eur. J., 2003, 9(5), 1166–1181 Search PubMed.
- ESI† contains additional details and includes calculations for isomeric structures of 41 and 42 as well as the energy profile from 43 to product.
- For a discussion of piano-stool iron complexes: (a) P. Buchgraber, L. Toupet and V. Guerchias, Organometallics, 2003, 22(24), 5144–5147 CrossRef CAS; (b) J. Zheng, L. C. Misal Castro, T. Roisnel, C. Darcel and J.-B. Sortais, Inorg. Chim. Acta, 2012, 380, 301–307 CrossRef CAS , Young Investigators Special Issue; (c) F. Jiang, D. Bézier, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2011, 353(2–3), 239–244 CrossRef CAS; (d) A. K. Singh, P. Kumar, M. Yadav and D. S. Pandey, J. Organomet. Chem., 2010, 695(4), 567–573 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General reaction procedures and characterizations of the 1,1-disubstituted allenes and the iron metallacycles from 9, 10, 11, and 23. Experimental procedures and characterization data for products 7, 8, 12–22, and 25. 1H NMR, 13C NMR and 2D NOESY spectra for all products, and X-ray crystal structure data for 18 and 24. A kinetic study of the reaction profile and a rate study which determines the order of the reaction with respect to alkyne concentration. Computational information regarding structure of the iron metallacycle, regiochemistry of Fe(CO)4 complexation, energies of possible intermediates in the mechanistic pathway, HOMO and LUMO calculations and exploration of the triplet potential energy surface. Also included are calculations of alternative structures of comparisons (S1–S145). CCDC 904093 and 824471. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21404k |
| This journal is © The Royal Society of Chemistry 2013 |