Palladium-catalyzed cascade reactions of 3-iodochromones with aryl iodides and norbornadiene leading to annulated xanthones†
Ming
Cheng
,
Jianwei
Yan
,
Feng
Hu
,
Hong
Chen
and
Youhong
Hu
*
State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 ZuChongZhi Road, 201203, Shanghai, China. E-mail: yhhu@mail.shcnc.ac.cn; Fax: +86 21-5080-5896
First published on 28th September 2012
Abstract
An efficient, palladium-catalyzed cascade reaction, which leads to formation of annulated xanthones, was devised. The process, which uses readily available 3-iodochromones, aryl iodides and norbornadiene as starting materials, takes place via a tandem Heck reaction/double C–H activation/retro-Diels–Alder pathway. The high chemoselectivity of the process is mechanistically unique and it serves as a new approach to achieve regioselective control of C–H activation in Pd/norbornene or norbornadiene systems. Its broad substrate scope, including heteroaryl coupling partners, enables access to diverse annulated xanthones.
Introduction
Among the important group of C–H activation/C–C bond forming processes, the Pd-catalyzed/norbornene-mediated reaction (Catellani reaction) is widely recognized as being a powerful method to form diversely functionalized aromatic compounds and condensed heterocycles. The Catellani reaction follows a cascade pathway in which C–C bonds and C–O or C–N bonds form sequentially.1 Generally, the “ortho effect” of aryl iodides ensures that Csp2–Csp2 rather than Csp2–Csp3 coupling of Pd(IV) intermediates occurs regioselectively to generate biaryl linkages and an active Pd(II) species, which participates in a variety of coupling reactions.2A large effort has been devoted to improving the efficiency and practicality of this Csp2–Csp2 coupling process.3 In contrast, the development of techniques to selectively promote Csp2–Csp3 reductive elimination of the key aryl Pd intermediate in the reaction pathway still presents a distinct challenge. In an early report, Catellani suggested that it might be possible to facilitate aryl–norbornyl bond formation by Csp2–Csp3 coupling when an ortho group is absent.4 Recently, Catellani and Malacria discovered that certain chelating functional groups on the second ortho substituent can guide alternate reactivity, thus offsetting the normal ortho effect.5 In the study described below, we have uncovered a new, palladium-catalyzed cascade reaction of 3-iodochromones with aryl iodides and norbornadiene that forms annulated xanthones via a Heck coupling/double C–H activation/retro-Diels–Alder sequence and that involves an unexpected Csp2–Csp3 reductive elimination reaction of the key aryl Pd intermediate even though no ortho chelating functional groups are present.
The chromone ring system is an important structural motif frequently found in natural products, pharmaceuticals and other important synthetic substances.6 The results of recent investigations have shown that C–H functionalization reactions could occur at the C-2 and C-3 position of chromones.7 In a continuation of an effort aimed at the development of strategies for the preparation of diverse natural-product-like scaffolds,8 we speculated that regioselective C–H activation at the C-2 position of chromones might take place in the course of the Catellani reaction and that this pathway would serve as a facile method for forming flavones and/or xanthones (Scheme 1).
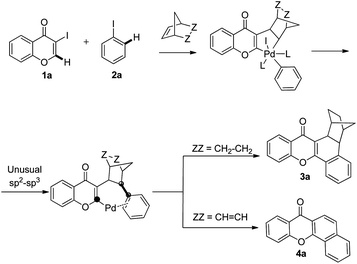 | ||
| Scheme 1 A proposed sp2–sp3 coupling process in the palladium-catalyzed reaction of 3-iodochromone with iodobenzene and norbornadiene or norbornene. | ||
Results and discussion
Initial studies aimed at exploring this proposal focused on the Pd-catalyzed reaction of 3-iodochromone (1a) with iodobenzene (2a) in the presence of norbornene. Under the reaction conditions described by Catellani (see procedure A), the unexpected product 3a is produced in 82% yield (Scheme 2). We hypothesized that this catalytic reaction proceeds via a pathway involving Csp2–Csp3 reductive elimination. Intrigued by this hypothesis, norbornadiene was utilized as a substrate in a reaction carried out under otherwise identical conditions. We believed that retro-Diels–Alder reaction would occur on the initially formed product to afford a benzoxanthone derivative. Importantly, substances in the benzoxanthone family display interesting bioactivities9 and, as a result, various methods for their synthesis have been described.10 However, the cascade process we envisioned would represent the most direct and functionally tolerant method for forming these targets (Table 1).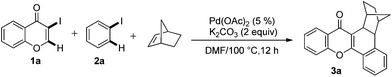 | ||
| Scheme 2 Pd-catalyzed tandem formation of compound 3a. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Pd cat. | Base | T/°C | t/h | Yieldd (%) |
a All reactions were carried out using the following molar ratio of the reagents: 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a:norbornadiene = 1:1:2.
b Freshly distilled DMF.
c Freshly distilled DMF with 1.5 equiv. of water.
d Isolated yield.
e The reaction was run with molar ratio of the reagents: 1a:2a:norbornadiene = 1:2:6. :2a:norbornadiene = 1:1:2.
b Freshly distilled DMF.
c Freshly distilled DMF with 1.5 equiv. of water.
d Isolated yield.
e The reaction was run with molar ratio of the reagents: 1a:2a:norbornadiene = 1:2:6.
|
||||||
| 1a | DMF | Pd(OAc)2 | K2CO3 | 90 | 12 | 10 |
| 2a | DMF | Pd(OAc)2 | Cs2CO3 | 90 | 12 | Messy |
| 3a | DMF | Pd(OAc)2 | CsPiv | 90 | 12 | 45 |
| 4a | DMA | Pd(OAc)2 | CsPiv | 90 | 12 | 40 |
| 5a | DMSO | Pd(OAc)2 | CsPiv | 90 | 12 | 31 |
| 6a | DMFb | Pd(OAc)2 | CsPiv | 90 | 12 | 18 |
| 7a | DMFc | Pd(OAc)2 | CsPiv | 90 | 12 | 50 |
| 8a | DMFc | Pd(OAc)2 | CsPiv | 130 | 3 | 57 |
| 9e | DMFc | Pd(OAc)2 | CsPiv | 130 | 3 | 78 |
| 10e | DMFc | PdCl2 | CsPiv | 130 | 3 | 56 |
| 11e | DMFc | Pd2(dba)3 | CsPiv | 130 | 3 | 47 |
| 12e | DMFc | PdCl2(PPh3)2 | CsPiv | 130 | 3 | 45 |
| 13e | DMFc | Pd(PPh3)4 | CsPiv | 130 | 3 | 38 |
| 14e | DMFc | PdCl2(PPh3)2 | CsPiv | 130 | 3 | 33 |
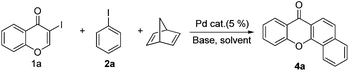
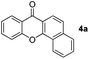
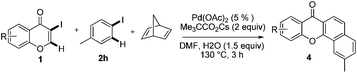
Indeed, reaction of 1a and 2a in the presence of norbornadiene, promoted by Pd(OAc)2 in the presence of K2CO3 (see procedure A), was observed to generate the desired benzoxanthone 4a in only 10% yield (entry 1). The low yield of the reaction might be a consequence of the high reactivity of norbornadiene.5a,11 When the softer base Cs2CO3 was utilized (entry 2), this reaction produces an inseparable mixture of products. Since the use of pivalate anion as a ligand is known to improve the efficiency of many C–H activation processes.12 Me3CCO2Cs (entry 3) was employed as a base in the reaction. Significantly, benzoxanthone 4a is produced in 45% yield under these conditions. Moreover, other higher boiling solvents, such as DMSO and DMA, could also be used to facilitate the final retro-Diels–Alder step (entries 4–6). However, freshly distilled DMF (entry 6) was not an effective solvent, indicating that water is required to promote the reaction.3g,k,13 The results of a brief examination showed that 1.5 equiv. of water is ideal. As expected, the reaction occurs more rapidly at higher temperatures (entry 8). Importantly, increasing the amounts of both iodobenzene and norbornadiene leads to a dramatically improved 78% yield (entry 9). Finally, although other common Pd sources can be utilized to promote this reaction, Pd(OAc)2 is the most efficient catalyst (entries 10–14). The combined studies demonstrate that DMF containing 1.5 equiv. H2O as the solvent at 130 °C, Me3CCO2Cs as the base, 5 mol% Pd(OAc)2 as the catalyst, 2 equiv. of PhI, 6 equiv. of norbornadiene, and 3 h reaction time are the optimal conditions for the new reaction.
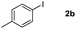
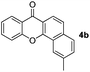
To gain insight into the mechanism of the process, reaction of 1a with para-iodotoluene (2b) was carried out under the optimized conditions. This reaction generates the benzoxanthone 4b in 78% yield. The structure of 4b and in particular the position of the methyl group was assigned by using X-ray crystallographic analysis (ESI†). Importantly, the position of methyl group in 4b is fully consistent with the proposed catalytic cycle, where Csp2–Csp3 rather than Csp2–Csp2 coupling takes place in the reductive elimination step (Scheme 3). The overall route could be initiated by oxidative addition of Pd(0) to 1a, followed by norbornadiene insertion, and intramolecular sp2 C–H activation at the 2-position of the chromone ring then forms the chromone fused palladacycle B. A second oxidative addition of the aryl iodide affords the Pd(IV) complex C, which then undergoes reductive elimination by either Csp2–Csp3 or Csp2–Csp2 coupling. The position of the methyl group in 4b shows that Csp2–Csp3 reductive elimination regioselectively gives intermediate D, which undergoes the second sp2 C–H activation to form F followed by release of the Pd(0) complex. The retro-Diels–Alder reaction of F, taking place at 130 °C leads to formation of the benzoxanthone product. An alternative mechanism may involve initial oxidative addition by ArI, which reacts with norbornadiene to generate a phenyl fused palladacycle and followed by a second oxidative addition of 3-iodochromone to give the same Pd(IV) complex C. Since 3-iodochromone with an electron-withdrawing group is more reactive than iodobenzene for the initial oxidative addition of Pd(0), the first possibility is favored. Also, we speculate that the selective operation of Csp2–Csp3 coupling is a consequence of stabilization of intermediate D by the electron-donating effect of the oxovinyl system.
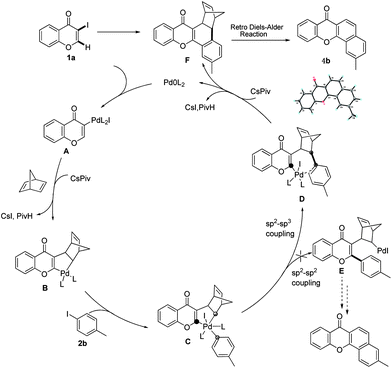 | ||
| Scheme 3 Proposed catalytic cycle. | ||
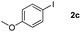
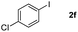
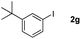
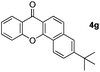
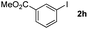
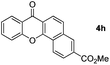
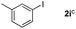
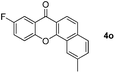
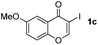
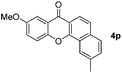
Next, differently substituted aryl iodides were used to explore the scope of the new tandem reaction, conducted under optimized conditions. The results, summarized in Table 2, show that the electronic nature of para-substituents on the aryl iodide has a significant effect on the reaction (entries 2–5). Specifically, electron donating group substituted aryl iodides undergo inefficient reactions owing to a slow oxidative insertion step converting Pd(II) to Pd(IV) (entry 3). In addition, the steric bulk of meta aryl iodide substituents can be used to govern the regioselectivity of the C–H activation step. As demonstrated by reactions of the meta-tert-butyl and -methoxycarbonyl substituted aryl iodides 2g and 2h that yield the respective benzoxanthones 4g and 4h, the course of the process is guided by steric hindrance (entries 7 and 8). As expected, mixtures of two benzoxanthones, arising by C–H activation at either ortho-phenyl site, are generated in reactions of the aryl iodides 2i, 2j and 2k, which contain smaller electron-donating or electron-withdrawing meta-groups (entries 9–11). It should be noted that reactions of aryl iodides bearing ortho-CH3, -OMe and -CF3 substituents are low yielding, perhaps because of steric effects that slow oxidative insertion in the Pd(II) intermediate B.


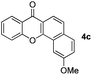
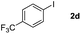
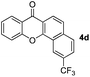
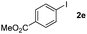
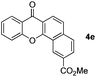
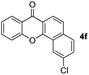
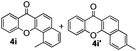
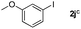
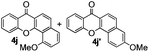
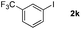
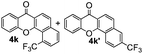
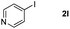
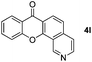
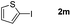
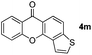

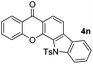
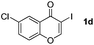
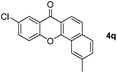
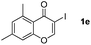
It is significant that the heteroaryl iodides, 2-iodothiophene, 4-iodopyridine and 3-iodoindole, undergo this reaction to give the desired products in moderate to good yields. In contrast, bromobenzene can not be used in place of its iodide counterpart as the coupling partner, presumably because it does not react with the palladium(II) metallacycle intermediate. Lastly, different 3-iodochromones containing both electron-withdrawing and -donating substituents on the chromone ring react with 4-iodotoluene (Table 3) to give the corresponding benzoxanthones in good yields.
|
|
|||
|---|---|---|---|
| Entry | Substrate 1 | Product | Yield (%) |
| 1 |

|
|
71 |
| 2 |
|
|
85 |
| 3 |
|
|
60 |
| 4 |
|
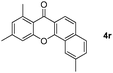
|
73 |
| 5 |

|
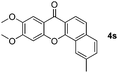
|
80 |
Conclusions
In the investigation described above, we have developed a highly efficient cascade process that involves unique C–H activation of the 2-position of the chromone ring system. The compatibility of this novel reaction with aryl iodides allows for an efficient method to prepare benzoxanthones. The high selectivity of the novel Pd/norbornene catalytic system should be applicable to reactions that result in the construction of interesting fused ring systems. Moreover, observations made in this study suggest that slightly different catalysts might be needed for reactions of each substrate type and that the Pd/norbornene catalytic system may be more mechanistically complicated than previously believed. We anticipate that the novel reaction discovered in this investigation will be applicable to other substrates and that future studies in this area will lead to the development of more comprehensive catalytic systems.Acknowledgements
This work was supported by grant from National Natural Science Foundation of China (21172232) and the Ministry of Science and Technology of China (2009CB940903)Notes and references
- For selected reviews, see: (a) M. Catellani, Top. Organomet. Chem., 2005, 14, 21 CAS; (b) M. Catellani, E. Motti and N. Della Ca, Acc. Chem. Res., 2008, 41, 1512 CrossRef CAS; (c) M. Catellani, E. Motti, F. Faccini and R. Ferraccioli, Pure Appl. Chem., 2005, 77, 1243 CrossRef CAS; (d) M. Lautens, D. Alberico, C. Bressy, Y.-Q. Fang, B. Mariampillai and T. Wilhelm, Pure Appl. Chem., 2006, 78, 351 CrossRef CAS; (e) A. Martins, B. Mariampillai and M. Lautens, Top. Curr. Chem., 2010, 292, 1 CrossRef CAS; (f) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS; (g) K. Muñiz, Angew. Chem., Int. Ed., 2009, 48, 9412 CrossRef; (h) P. Sehnal, R. J. K. Taylor and I. J. S. Fairlamb, Chem. Rev., 2010, 110, 824 CrossRef CAS.
- (a) G. Maestri, E. Motti, N. Della Ca, M. Malacria, E. Derat and M. Catellani, J. Am. Chem. Soc., 2011, 133, 8574 CrossRef CAS; (b) M. Catellani and E. Motti, New J. Chem., 1998, 22, 759 RSC.
- For selected recent examples of the Catellani reaction via sp2–sp2 coupling, see: (a) M. Blanchot, D. A. Candito, F. Larnaud and M. Lautens, Org. Lett., 2011, 13, 1486 CrossRef CAS; (b) D. A. Candito and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 6713 CrossRef CAS; (c) N. Della Ca, G. Maestri and M. Catellani, Chem.–Eur. J., 2009, 15, 7850 CrossRef CAS; (d) N. Della Ca, E. Motti, A. Mega and M. Catellani, Adv. Synth. Catal., 2010, 352, 1451 CrossRef CAS; (e) K. M. Gericke, D. I. Chai, N. Bieler and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 1447 CrossRef CAS; (f) F. Jafarpour and H. Hazrati, Adv. Synth. Catal., 2010, 352, 363 CrossRef CAS; (g) L. Jiao and T. Bach, J. Am. Chem. Soc., 2011, 133, 12990 CrossRef CAS; (h) G. Maestri, N. Della Ca and M. Catellani, Chem. Commun., 2009, 4892 RSC; (i) G. Maestri, M.-H. Larraufie, E. Derat, C. Ollivier, L. Fensterbank, E. Lacôte and M. Malacria, Org. Lett., 2010, 12, 5692 CrossRef CAS; (j) E. Motti, N. Della Ca, S. Deledda, E. Fava, F. Panciroli and M. Catellani, Chem. Commun., 2010, 46, 4291 RSC; (k) Y.-B. Zhao, B. Mariampillai, D. A. Candito, B. Laleu, M. Li and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 1849 CrossRef CAS; (l) D. G. Hulcoop and M. Lautens, Org. Lett., 2007, 9, 1761 CrossRef CAS.
- (a) K. Albrecht, O. Reiser, M. Weber, B. Knieriem and A. de Meijere, Tetrahedron, 1994, 50, 383 CrossRef CAS; (b) M. Catellani and G. P. Chiusoli, J. Organomet. Chem., 1985, 286, c13 CrossRef CAS.
- (a) N. Della Ca, G. Maestri, M. Malacria, E. Derat and M. Catellani, Angew. Chem., Int. Ed., 2011, 50, 12257 CrossRef CAS; (b) M.-H. Larraufie, G. Maestri, A. Beaume, É. Derat, C. Ollivier, L. Fensterbank, C. Courillon, E. Lacôte, M. Catellani and M. Malacria, Angew. Chem., Int. Ed., 2011, 50, 12253 CrossRef CAS.
- (a) S. Khadem and R. J. Marles, Molecules, 2011, 17, 191 CrossRef; (b) N. F. L. Machado and M. P. M. Marques, Curr. Bioact. Compd., 2010, 6, 76 CrossRef CAS; (c) S. K. Sharma, S. Kumar, K. Chand, A. Kathuria, A. Gupta and R. Jain, Curr. Med. Chem., 2011, 18, 3825 CrossRef CAS.
- (a) D. Kim and S. Hong, Org. Lett., 2011, 13, 4466 CrossRef CAS; (b) F. Chen, Z. Feng, C. Y. He, H.-Y. Wang, Y. L. Guo and X. Zhang, Org. Lett., 2012, 14, 1176 CrossRef CAS; (c) K. H. Kim, H. S. Lee, S. H. Kim and J. N. Kim, Tetrahedron Lett., 2012, 53, 2761–2764 CrossRef CAS; (d) M. Khoobi, M. Alipour, S. Zarei, F. Jafarpour and A. Shafiee, Chem. Commun., 2012, 48, 2985–2987 RSC.
- (a) B. Chao, S. Lin, Q. Ma, D. Lu and Y. Hu, Org. Lett., 2012, 14, 2398 CrossRef CAS; (b) H. Chen, F. Xie, J. Gong and Y. Hu, J. Org. Chem., 2011, 76, 8495 CrossRef CAS; (c) L. Zhao, F. Xie, G. Cheng and Y. Hu, Angew. Chem., Int. Ed., 2009, 48, 6520 CrossRef CAS; (d) F. Xie, H. Chen and Y. Hu, Org. Lett., 2010, 12, 3086 CrossRef CAS; (e) Y. Liu, L. Huang, F. Xie and Y. Hu, J. Org. Chem., 2010, 75, 6304 CrossRef CAS; (f) Y. Liu, L. Huang, F. Xie, X. Chen and Y. Hu, Org. Biomol. Chem., 2011, 9, 2680 RSC.
- (a) H.-J. Cho, M.-J. Jung, S. Woo, J. Kim, E.-S. Lee, Y. Kwon and Y. Na, Bioorg. Med. Chem., 2010, 18, 1010 CrossRef CAS; (b) Y. Ren, L. B. S. Kardono, S. Riswan, H. Chai, N. R. Farnsworth, D. D. Soejarto, E. J. Carcache de Blanco and A. D. Kinghorn, J. Nat. Prod., 2010, 73, 949 CrossRef CAS; (c) G. W. Rewcastle, G. J. Atwell, L. Zhuang, B. C. Baguley and W. A. Denny, J. Med. Chem., 1991, 34, 217 CrossRef CAS.
- (a) N. Barbero, R. SanMartin and E. Dominguez, Tetrahedron, 2009, 65, 5729 CrossRef CAS; (b) N. Barbero, R. SanMartin and E. Dominguez, Green Chem., 2009, 11, 830 RSC; (c) E. Chevenier, C. Lucatelli, U. Pandya, W. Wang, Y. Gimbert and A. E. Greene, Synlett, 2004, 2693 CAS; (d) D. S. Clarke, C. D. Gabbutt, J. D. Hepworth and B. M. Heron, Tetrahedron Lett., 2005, 46, 5515 CrossRef CAS; (e) S. H. Kim, J. R. Gunther and J. A. Katzenellenbogen, Org. Lett., 2008, 10, 4931 CrossRef CAS; (f) R. C. Larock and Q. Tian, J. Org. Chem., 1998, 63, 2002 CrossRef CAS; (g) J. Li, C. Jin and W. Su, Heterocycles, 2011, 83, 855 CrossRef CAS; (h) W.-Z. Xu, Z.-T. Huang and Q.-Y. Zheng, J. Org. Chem., 2008, 73, 5606 CrossRef CAS; (i) J. Zhao and R. C. Larock, Org. Lett., 2005, 7, 4273 CrossRef CAS; (j) J. Zhao and R. C. Larock, J. Org. Chem., 2007, 72, 583 CrossRef CAS.
- (a) E. Motti and M. Catellani, Adv. Synth. Catal., 2008, 350, 565 CrossRef CAS.
- (a) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848 CrossRef CAS; (b) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS; (c) M. Lafrance, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 14570 CrossRef CAS; (d) M. Nakanishi, D. Katayev, C. Besnard and E. P. Kündig, Angew. Chem., Int. Ed., 2011, 50, 7438 CrossRef CAS.
- S. Deledda, E. Motti and M. Catellani, Can. J. Chem., 2005, 83, 741 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 884029 (4b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21335d |
| This journal is © The Royal Society of Chemistry 2013 |