(NHCMe)SiCl4: a versatile carbene transfer reagent – synthesis from silicochloroform†
Tobias
Böttcher
,
Bassem S.
Bassil
,
Lyuben
Zhechkov
,
Thomas
Heine
and
Gerd-Volker
Röschenthaler
*
School of Engineering and Science, Jacobs University Bremen, Campus Ring 1, D-28759 Bremen, Germany. E-mail: g.roeschenthaler@jacobs-university.de; Tel: +49 421 200-3138
First published on 14th September 2012
Abstract
A new synthetic pathway for the N-heterocyclic carbene adduct (NHCMe)SiCl4 (2) (NHCMe = 1,3-dimethylimidazolidin-2-ylidene) using silicochloroform is presented. Supported by DFT calculations, the energy for dissociation of 2 into the carbene and the SiCl4 fragment was found to be comparable to carbene transfer reagents based on silver(I) chloride. Compound 2 was used to transfer the NHC ligand to three different phosphorus(III) chloro compounds, resulting in the neutral complexes (NHCMe)PCl3 (3a), (NHCMe)PCl2Ph (3b) and (NHCMe)PCl2Me (3c). The sterically non-demanding NHC ligand allowed the phosphorus(III) in complex 3a to be oxidized to phosphorus(V) without loss of the NHC ligand, and afford (NHCMe)PF4H (4). Furthermore, bis-carbene complexes of Ni(II) (5) and Pd(II) (6) were obtained by reacting 2 with the respective metal chlorides.
Introduction
N-heterocyclic carbene (NHC) complexes have been of great interest to chemists since the 1960s, although the first stable NHC was only isolated in 1991.1 Since then, free and stable NHCs have been available for preparative chemistry, and many coordination compounds of metals and non-metals coordinated by NHCs have been reported.2 Later on, Herrmann et al. reported the catalytic property of palladium NHC complexes, making the research of NHC-coordinated transition metals one of the fastest growing topics in coordination chemistry ever since.3 During the last two decades, numerous reports in literature on the application of NHC complexes in metal-mediated catalysis, organocatalysis, pharmaceutical applications and technical processes have appeared.4 Although carbenes have generally become an established class of ligands, their synthesis and isolation, either in the free form or as part of a complex, is an ongoing challenge. The high stability of the NHC–transition-metal bond provides effective pathways in the synthesis of the resulting complex, including ligand exchange, in situ deprotonation of imidazolium, oxidative addition to halo-imidazolium, and transmetallation.5However, the coordination of NHC to main-group elements is more constrained. Very recently, there have been remarkable advances in the chemistry of p-block compounds, where the coordination by NHCs resulted in unusual bonding properties and oxidation states. Bertrand and co-workers isolated a series of NHC-stabilized phosphorus(0) compounds by addition of different free NHCs to white phosphorus.6 Robinson and co-workers reduced NHCdipp (NHCdipp = :C{N(2,6-iPr2C6H3)CH}2) complexes of silicon(IV) in order to form Si–Si single and double bonds. Reduction of (NHCdipp)SiCl4 resulted in the first complex with silicon being in the formal oxidation state of zero, (NHCdipp)Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si(NHCdipp).7 Following the same procedure, they reduced (NHCdipp)PCl3 to give (NHCdipp)PP(NHCdipp), as well as (NHCdipp)AsCl3 to give (NHCdipp)AsAs(NHCdipp).8 Jones et al. followed a modified procedure to synthesize (NHCdipp)GeGe(NHCdipp) by reduction from (NHCdipp)GeCl2.9 In addition to these diatomic allotropes, other compounds of main-group element species stabilized by N-heterocyclic carbenes were also synthesized.10
Si(NHCdipp).7 Following the same procedure, they reduced (NHCdipp)PCl3 to give (NHCdipp)PP(NHCdipp), as well as (NHCdipp)AsCl3 to give (NHCdipp)AsAs(NHCdipp).8 Jones et al. followed a modified procedure to synthesize (NHCdipp)GeGe(NHCdipp) by reduction from (NHCdipp)GeCl2.9 In addition to these diatomic allotropes, other compounds of main-group element species stabilized by N-heterocyclic carbenes were also synthesized.10
The starting compounds for the above mentioned reduction reactions as well as most other adducts of NHCs to main-group elements are prepared starting from free, uncoordinated NHCs. This approach imposes the use of NHCs with bulky substituents at the N,N′-positions, as they provide steric protection to the carbene center and hence stability to the NHCs. Moreover, these substituents allowed the isolation of the diatomic allotropes mentioned above. On the other hand, these bulky groups do not allow any additional reaction at the coordinated center of the aforementioned NHC complexes because of steric hindrance. The challenge lies therefore in a synthetic pathway, which does not require the presence of sterically demanding NHC ligands, in order to allow further reactions at the carbene-coordinated center and to have a NHC-containing starting material stable enough to be isolated but active enough to allow the NHC ligands to undergo further coordination.
Herein we report the synthesis of the NHC transfer reagent (NHCMe)SiCl4 (2, NHCMe = 1,3-dimethylimidazolidin-2-ylidene) and its reactivity towards main-group and transition-metal elements. We chose NHCMe as it is the smallest imidazolidine-based NHC and has not yet been isolated in its free form, unlike its unsaturated analog.11 We performed ligand transfer reaction on 2 in order to synthesize adducts of NHCMe and phosphorus(III) as well as nickel(II) and palladium(II). We also proved the accessibility of the carbene-coordinated phosphorus(III) by oxidizing it to phosphorus(V) without loss of NHCMe. As a source of silicon we first used hexachlorodisilane and then silicochloroform, which is a raw material in chemical industry, providing precursor 2 very inexpensively.12
Results and discussion
Synthesis of (NHCMe)SiCl4
The reaction of 2-chloro-1,3-dimethylimidazolinium chloride (1) with hexachlorodisilane in dichloromethane gave (NHCMe)SiCl4 (2) in good yield. The mechanism depicted in Scheme 1 as ‘route A’ proposes the initial addition of chloride to hexachlorodisilane, which leads to Si–Si bond cleavage and formation of silicon tetrachloride and trichlorosilanide. The latter species is a very reactive nucleophile and attacks the carbon at the 2-position in 2-chloroimidazolinium. The chlorine is then abstracted by the Lewis-acidic silicon and 2 eventually is formed by redox-rearrangement. The cleavage of hexachlorodisilane by bases was described by Wilkins in 1953 and has been since then used for trichlorosilylation reactions.13 Our group recently introduced 2,2-difluoro-1,3-dimethylimidazolidine, which can be considered as the fluorine analog of 1, as a carbene precursor for main-group fluoro-complexes.14 We also proposed a mechanism involving the formation of a phosphoranide by abstraction of one of the fluorides, which then attacks the carbon at the 2-position, followed by redox-rearrangement which leads to (NHCMe)PF5.14a This is in agreement with the proposed mechanism depicted in Scheme 1. In the case of silicon, there is no neutral analog to PF3, although hexachlorodisilane has just been recently discussed as a source for :SiCl2.15 Other sources of trichlorosilanide include silicochloroform, which has been extensively investigated by Benkeser and others.16 Consequently, the reaction of 1 with silicochloroform in presence of triethylamine also yields to product 2 (route B in Scheme 1).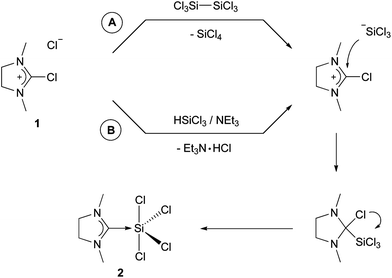 | ||
| Scheme 1 Synthesis of 2. | ||
Compound 2 is a colorless, non-hygroscopic solid which is stable under an inert atmosphere. It is sensitive towards air and moisture, but can be handled using standard Schlenk-techniques without the need of a glovebox. It is soluble in acetonitrile, dichloromethane and tetrahydrofuran, sparingly soluble in toluene, and insoluble in methyl tert-butyl ether, diethyl ether and alkanes. Complex 2 was fully characterized by multinuclear NMR spectroscopy, elemental analysis and XRD. Single crystals suitable for X-ray diffraction were grown by slow diffusion of diethyl ether into a solution of 2 in acetonitrile. Compound 2 crystallizes in the orthorhombic space group Pbca; the structure corresponds to silicon(IV) in a trigonal-bipyramidal geometry, with the NHC ligand at the equatorial position (Fig. 1). This arrangement is consistent with known derivatives based on SiCl4 (Scheme 2) and SiBr4.7,17 The only structural exception known so far, in which the NHC ligand occupies the axial position, is the fluorine analog recently reported by Roesky and co-workers.17a
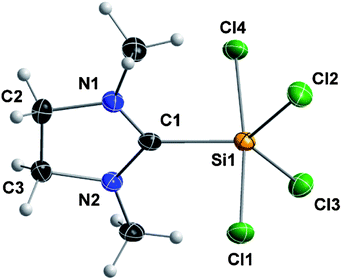 | ||
| Fig. 1 Crystal structure of compound 2 with the thermal ellipsoids set at 50% probability level. Selected bond lengths (pm) and angles (°): Si1–C1 192.8(3), Si1–Cl1 220.22(12), Si1–Cl2 207.27(11), Si1–Cl3 207.32(12), Si1–Cl4 221.39(12); N1–C1–N2 110.4(3), Cl1–Si1–Cl4 172.95(6), Cl2–Si1–Cl3 113.54(5). | ||
 | ||
| Scheme 2 Synthesis of known analogs of 2. | ||
In the solid-state structure of 2, the C1–Si1 bond (192.8(3) pm) is slightly longer than that in B1 (191.1(7) pm) and almost identical to that in B2 (192.8(2) pm), no crystal structure was reported for B3. Although 2 has a saturated NHC backbone, the angular sum for each nitrogen is 360° and the ring is almost planar (deviation from the N1–C1–N2 plane is −6.5 pm for C3 and 7.6 pm for C2).
The 29Si NMR singlet of 2 (−103.9 ppm, CD3CN) shows a chemical shift similar to that reported for B1 (−105.1 ppm, C6D6), B2 (−108.9 ppm, CD2Cl2) and B3 (−110.1 ppm, C4D8O) and is more upfield than SiCl4 (−18.5 ppm, CCl4).18 Furthermore, its carbene 13C NMR signal (173.1 ppm, CD3CN) is more downfield compared to B1 (153.1 ppm, C6D6) and B3 (156.9 ppm, C4D8O) (no 13C NMR data for B2 is available), which is expected for a saturated NHC complex.19
Quantum mechanical models of (NHCMe)SiCl4
Stability and structures of carbene-derived penta- and hexacoordinated silicon complexes have been studied earlier.20 However, here we focus on a peculiarity of the binding between the SiCl4 and carbene fragments. Recently, Liu studied the bonding situation for compound C, as a simplified model of Robinson's compound B2, making it the unsaturated analog of our compound 2 (Fig. 2).21 He applied an energy decomposition analysis (EDA)22 on C, using DFT (BP86 functional and frozen core TZ2P basis set).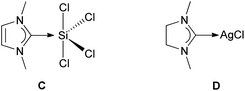 | ||
| Fig. 2 Model compounds C and D. | ||
Liu came to three conclusions: (i) the formation of the compound from SiCl4 and the carbene fragment should not follow the formation of a classical Lewis-acid/Lewis-base adduct, (ii) the Si–C bond is based on purely electrostatic interaction, (iii) the Si–C bond is highly polarized. As the binding between the SiCl4 and the carbene fragment is of most importance for the chemistry presented further in this article, we have carried out additional calculations to complement those performed by Liu.
As a method of choice, we have employed DFT23 calculations using PBE24 functional and full electron TZ2P ZORA25 basis set with scalar relativistic correction, as implemented in adf2012.01.26 We have performed three type of analyses: AIM27, NBO28 and fragment analysis.29 AIM and NBO did not contribute new information. The fragment and EDA analysis, however, revealed that binding between the two fragments (NHC and SiCl4) is manifested not only by the electrostatic interaction. Although it is the predominant term, significant stabilization is due to the orbital interactions, which suggest that the binding between the fragments indeed has substantial donor–acceptor character (Table S5, ESI†). The fragment analysis also indicates that part of the energy stabilization of the binding between the two fragments is due to interactions of molecular orbitals which have very little or no contribution from the atoms forming the C–Si bond (see Fig. 3).
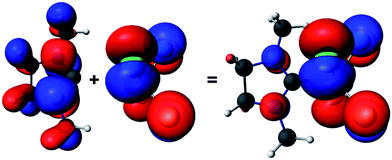 | ||
| Fig. 3 Fragment molecular orbitals of 2: NHC fragment (left, HOMO−1 for this fragment), SiCl4 fragment (middle, HOMO for this fragment) and the resulting HOMO orbital of 2 (right). The isosurfaces are drawn at 0.03 a.u. | ||
According to the fragment analysis, the HOMO of 2 can be represented as emerging from mainly two fragment molecular orbitals (FMOs), each contributing with different weight. The two FMOs and the resulting HOMO of 2 are depicted in Fig. 3 (Fig, S27, ESI†).
The SiCl4 FMO is delocalized only on the four Cl atoms of the fragment and constitutes about 90% of the HOMO in compound 2. This leads to the conclusion that a chemical attack or deformation of the SiCl4 fragment would result in destabilization of the interaction between the two moieties.
A clear and straightforward way to assess the effect is to compute the binding energy between the fragments for a model where the SiCl4 would have an artificial geometrical configuration. An example is where the four Cl atoms lie in one plane with the Si atom (2*). The transformation from 2 to 2* has a penalty of 15 kcal mol−1 on the binding energy between the two fragments. Thus, the bond dissociation energy for the activated state 2* becomes similar to that found for NHC–AgCl (compound D, Fig. 2), which we included as a model for widely-used NHC transfer reagents.
The bond dissociation energies (BDE) of 2, 2*, C and D (Table 1) revealed almost the same value for 2 (68 kcal mol−1) and C (69 kcal mol−1), even though the NHC of C is an unsaturated ring system. Unsaturated five-membered NHCs do not exhibit full π-delocalization, but rather two separated π-systems.30 In this case, the double bond in C has a negligible influence on the silicon–carbene bond. The decrease in BDE going from 2 to the activated 2* indicates that NHC–SiCl4 can be used as an NHC transfer reagent. With these computational results we can conclude that the title compound 2 has a similar carbene transfer potential as the established carbene transfer reagent based on silver(I) (D).
| NBO charges | |||||
|---|---|---|---|---|---|
| Structure | BDE/kcal mol−1 | C | N1 | N2 | Si (Ag) |
| 2 | 68 | 0.156 | −0.291 | −0.291 | 1.108 |
| C | 69 | — | — | — | — |
| 2* | 53 | 0.194 | −0.303 | −0.300 | 1.079 |
| D | 55 | 0.094 | −0.318 | −0.319 | 0.516 |
NHC transfer to phosphorus(III)
The results presented above, as well as its straightforward and inexpensive accessibility through the synthesis using silicochloroform, led to the consideration of testing 2 as a reagent for carbene transfer. Addition of PCl3, PCl2Ph and PCl2Me to a solution of 2 in THF at 0 °C followed by stirring at room temperature for 12 h gave the NHCMe adducts (NHCMe)PCl3 (3a), (NHCMe)PCl2Ph (3b) and (NHCMe)PCl2Me (3c) in almost quantitative yields (Scheme 3).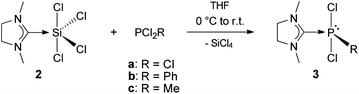 | ||
| Scheme 3 Synthesis of NHC–phosphorus(III) adducts 3a–c. | ||
Compounds 3a–c are colorless solids, which are stable under an inert atmosphere for at least several weeks. Furthermore, TGA measurements on these solids show decomposition between 70 and 110 °C (see ESI†). They are soluble in acetonitrile, dichloromethane, chloroform and THF, but insoluble in diethyl ether and alkanes. All compounds were characterized by multinuclear NMR, elemental analysis and XRD. The 31P NMR (CD3CN) shifts (24.6 ppm for 3a, −21.6 ppm for 3b and −22.1 ppm for 3c) are significantly upfield compared to the starting materials by around 200 ppm, due to electron donation of the NHC ligand. Robinson and co-workers have previously reported the analogous complex (NHCdipp)PCl3 (E) by addition of the respective free carbene to PCl3.8a,31 The 31P NMR upfield shift of E to 16.9 ppm (C6D6) is in agreement with our data. All 13C signals are split into doublets by the adjacent phosphorus and reveal no significant deviation in chemical shifts and coupling constants amongst each other. The respective spectra showed a shift of 172.9 ppm (1JCP = 109 Hz) for 3a, 174.9 ppm (1JCP = 78 Hz) for 3b and 176.1 ppm (1JCP = 82 Hz) for 3c. If kept in solution, decomposition of the three compounds to the parent phosphines can be detected by 31P NMR spectroscopy after one day. The respective 1H NMR shows decomposition products indicating a dissociation of NHC and phosphine, as already described for the reaction of the free carbene with chloroform.32
Adduct 3a crystallizes in the monoclinic space group P21/c, 3b in the monoclinic space group C2/c and 3c in the orthorhombic space group Pbca.
The three compounds are structurally analogous and reveal a pseudo-trigonal-bipyramidal geometry around phosphorus. The NHC ligand, the lone pair and the substituent R occupy the equatorial positions and the remaining two chlorine atoms reside at the axial positions (Fig. 4). The carbene–phosphorus bond lengths vary only slightly between the derivatives (187.87(16) pm for 3a, 187.07(15) pm for 3b and 187.64(16) pm for 3c) and are in good agreement with Robinson's compound E (187.1(11) pm).
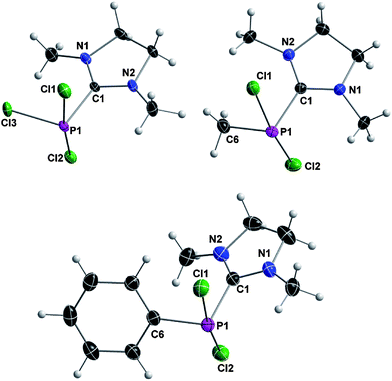 | ||
| Fig. 4 Crystal structures of 3a (top left), 3b (bottom) and 3c (top right) with thermal ellipsoids set at 50% probability level. For selected bond lengths and angles see Table 2. | ||
| 3a | 3b | 3c | |
|---|---|---|---|
| P1–C1 | 187.87(16) | 187.07(15) | 187.64(16) |
| P1–Cl1 | 223.32(6) | 241.61(6) | 246.91(7) |
| P1–Cl2 | 248.20(7) | 232.27(5) | 229.47(7) |
| P1–R | 205.37(6) | 184.82(15) | 184.35(18) |
| N1–C1–N2 | 111.72(14) | 111.17(13) | 111.11(14) |
| C1–P1–R | 103.22(5) | 105.75(7) | 104.47(7) |
| Cl1–P1–Cl2 | 170.66(2) | 171.88(2) | 172.24(2) |
Compound E was the only previous neutral adduct of carbene and phosphorus(III) chloride. Other reactions of sterically non-demanding NHCs with phosphorus(III) compounds gave only cationic products as shown in Scheme 4.33 The direct addition of free NHC to PCl3 led to the reduction of phosphorus and formation of F.34 Most likely, the free carbene attacks a chloride, instead of directly coordinating to phosphorus. A different approach was made by utilizing imidazolium-2-carboxylate (G), which has been used for synthesizing new transition-metal complexes.35 Reaction of G with substituted chlorophosphines afforded the cationic complexes H and I.36
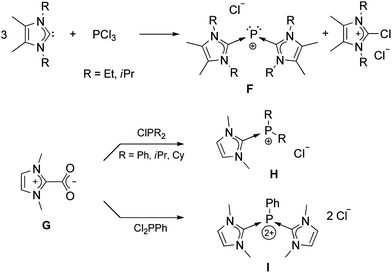 | ||
| Scheme 4 Cationic species resulting from the addition of NHC precursors to different phosphorus(III) compounds. | ||
It should also be noted that an analog of F was reported almost 30 years ago by Schmidpeter et al. by reaction of PCl3 with electron-rich bis-imidazolidinylidenes (“Wanzlick-Olefin”).37
For our NHC transfer reaction however, no cationic products were observed, and a proposed mechanism is shown in Scheme 5. Abstraction of a chloride from 2 by PCl2R leads to the imidazolinium-2-trichlorosilyl cation (J) and the phosphoranide [PCl3R]−.38 Cation J has not been isolated so far, however its bromine and methyl analogs were reported recently.39 The phosphoranide itself then undergoes a nucleophilic attack on cation J to form the transition-state assembly K according to Scheme 5. Loss of SiCl4 from K leads to the products 3a–c.
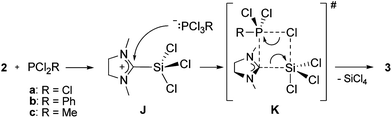 | ||
| Scheme 5 Proposed mechanism for the formation of 3a–c. | ||
In contrast to the phosphorus in E, which is protected towards oxidation by the sterically demanding NHCdipp ligand, the phosphorus in 3a is exposed and can easily be oxidized without loss of the carbene ligand. Addition of triethylamine trihydrofluoride to a cooled solution of 3a in dichloromethane, followed by an activation step with triethylamine, gave (NHCMe)PF4H (4) (Scheme 6).40 Chloride/fluoride metathesis occurs during the reaction, followed by addition of HF. Complex 4 is a colorless solid, which is soluble in acetonitrile and dichloromethane, but insoluble in diethyl ether and alkanes. It is stable towards air and moisture and can be washed with ethanol for purification.
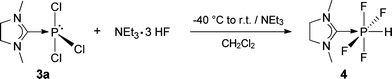 | ||
| Scheme 6 Chloride/fluoride metathesis and subsequent addition of HF to 3a. | ||
The 1H NMR (CD3CN) spectrum of 4 (Fig. 5) shows singlet signals at 3.20 ppm (6H) and 3.71 ppm (4H), which are assigned to the coordinated NHCMe ligand. At 5.72 ppm the P–H proton is detected as a doublet with a large coupling constant of 1JHP = 938 Hz. The doublet is split into quintets by the four adjacent fluorine atoms with a geminal coupling constant of 2JHF = 118 Hz.
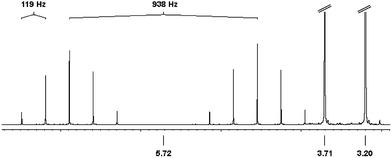 | ||
| Fig. 5 1H NMR spectrum of 4 in CD3CN. δ 3.20 (s, 6H, NCH3), 3.71 (s, 4H, –CH2–), 5.72 (d, quint, 1H, P–H,1JPH = 938 Hz, 2JHF = 119 Hz). (signals at δ 3.71 and 3.20 are cut off for clarity). | ||
Complex 4 was characterized by multinuclear NMR spectroscopy, HRMS and XRD. X-Ray quality single crystals were obtained after keeping a solution of 4 in acetonitrile for 7 days at −40 °C. Compound 4 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and the crystal structure reveals an octahedral geometry around phosphorus (Fig. 6). Our group recently reported analogs of 4 with the same octahedral geometry, in which hydrogen is replaced by fluorine, phenyl and methyl, via oxidative addition of 2,2-difluoro-1,3-dimethylimidazolidine to PF3, Cl2PPh and Cl2PMe, respectively.14a
and the crystal structure reveals an octahedral geometry around phosphorus (Fig. 6). Our group recently reported analogs of 4 with the same octahedral geometry, in which hydrogen is replaced by fluorine, phenyl and methyl, via oxidative addition of 2,2-difluoro-1,3-dimethylimidazolidine to PF3, Cl2PPh and Cl2PMe, respectively.14a
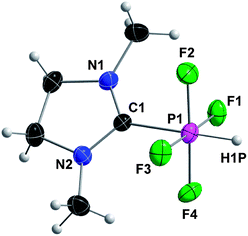 | ||
| Fig. 6 Crystal structure of 4 with thermal ellipsoids set at 50% probability level. Selected bond lengths (pm) and angles (°): C1–P1 190.6(2), av. P1–F 163.37(14), P1–H 134(2); N1–C1–N2 110.17(17), C1–P1–H 179.3(9), F1–P1–F3 178.00(7), F2–P1–F4 177.71(7). | ||
NHC transfer to nickel(II) and palladium(II)
Addition of solid Ni(PPh3)2Cl2 and PdCl2 to a solution of 2 in THF, followed by refluxing for 12 h, gave the bis-carbene complexes (NHCMe)2NiCl2 (5) and (NHCMe)2PdCl2 (6), respectively (Scheme 7). Both complexes are isostructural and crystallize in the monoclinic space group P21/n. The crystal structures reveal square-planar geometry around the metal centers with the NHC ligands in cis-conformation (Fig. 7).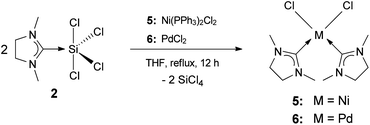 | ||
| Scheme 7 Synthesis of transition-metal complexes 5 and 6. | ||
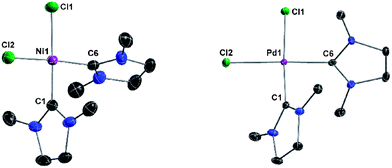 | ||
| Fig. 7 Crystal structures of 5 (left) and 6 (right) with thermal ellipsoids set at 50% probability level. H atoms are omitted for clarity. Selected bond lengths (pm) and angles (°) for 5: C1–Ni1 186.3(2), C6–Ni1 187.9(2), Ni1–Cl1 222.56(6), Ni1–Cl2 221.27(7); N1–C1–N2 108.8(2), N3–C6–N4 108.62(18), C1–Ni1–Cl1 174.24(7), C6–Ni1–Cl2 170.96(7). For 6: C1–Pd1 198.8(2), C6–Pd1 197.52(2), Pd1–Cl1 238.03(2), Pd1–Cl2 237.81(2); N1–C1–N2 109.2(2), N3–C6–N4 109.2(2), C1–Pd1–Cl1 176.28(7), C6–Pd1–Cl2 176.65(7). | ||
The two carbene ligands are twisted relative to the MCl2-plane with planar angles of 86.7 and 80.4° for 5 and 63.6 and 64.0° for 6. Complex 5 was reported by Lappert and Pyesome some time ago by addition of a nickel(0) precursor to bis(1,3-dimethylimidazoline) and subsequent oxidation with chlorine.41 Though, it has not been characterized in the solid state. Herrmann et al. reported the iodine analog of 6 with unsaturated NHCMe ligands by deprotonation of 1,3-dimethylimidazolium iodide with [Pd(OAc)2].3a
Conclusions
We have synthesized the adduct (NHCMe)SiCl4 (2) using silicochloroform and demonstrated its application as a versatile carbene transfer reagent. Direct addition of 2 to PCl3, PCl2Ph and PCl2Me in THF afforded the respective neutral (NHCMe)–phosphorus(III) complexes 3a–c in almost quantitative yields. Previous attempts to synthesize such compounds proved unsuccessful and only led to cationic species. Moreover, the phosphorus(III)-center in 3a is prone to oxidation, due to the sterically non-demanding NHCMe ligand. Without loss of the NHCMe ligand, complex 3a was oxidized to give (NHCMe)PF4H (4). The ligand NHCMe was also transferred from 2 to the transition metals nickel(II) and palladium(II) to give the bis-carbene complexes 5 and 6, respectively. All these compounds were fully characterized by single-crystal X-ray diffraction. The experimental results are supported by DFT calculations, which concluded a similar carbene transfer potential of 2 compared to established transfer reagents based on silver(I) chloride. The synthesis of 2 using silicochloroform makes it a low-cost alternative to the established yet expensive carbene transfer reagents based on silver.Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft (Grant RO 362/47-1) is gratefully acknowledged. Marie Curie (call: FP7-PEOPLE-2009-IAPP) QUASINANO. We thank Peter Brackmann (Universität Bremen, Germany) for X-ray crystallographic measurements.Notes and references
- (a) K. Öfele, J. Organomet. Chem., 1968, 12, P42–P43 CrossRef; (b) H. W. Wanzlick and H. J. Schönherr, Angew. Chem., Int. Ed. Engl., 1968, 7, 141–142 CrossRef CAS; (c) A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef.
- (a) W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef CAS; (b) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS; (c) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS; (d) P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef; (e) P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599–3611 CrossRef CAS; (f) J. C. Lin, R. T. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS; (g) N. Kuhn and A. Alsheikh, Coord. Chem. Rev., 2005, 249, 829–857 CrossRef CAS; (h) W. Kirmse, Eur. J. Org. Chem., 2005, 237–260 CrossRef CAS.
- (a) W. A. Herrmann, M. Elison, J. Fischer, C. Köcher and G. R. J. Artus, Angew. Chem., Int. Ed. Engl., 1995, 34, 2371–2374 CrossRef CAS; (b) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS; (c) C. S. J. Cazin, N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, Springer, Berlin, 2011 Search PubMed.
- (a) B. Alcaide, P. Almendros and A. Luna, Chem. Rev., 2009, 109, 3817–3858 Search PubMed; (b) K. J. Cavell and D. S. McGuinness, Coord. Chem. Rev., 2004, 248, 671–681 Search PubMed; (c) S. Diez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 Search PubMed; (d) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 Search PubMed; (e) F. Wang, L. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804–853 Search PubMed; (f) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 Search PubMed; (g) C. Samojlowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 Search PubMed; (h) K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 Search PubMed; (i) A. Kascatan-Nebioglu, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Coord. Chem. Rev., 2007, 251, 884–895 Search PubMed; (j) F. Glorius, N-Heterocyclic Carbenes in Catalysis, Springer, Berlin, 2007 Search PubMed; (k) N. Marion, S. Diez-Gonzalez and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 Search PubMed; (l) V. Nair, S. Bindu and V. Sreekumar, Angew. Chem., Int. Ed., 2004, 43, 5130–5135 Search PubMed.
- E. Peris, N-Heterocyclic Carbenes in Catalysis, Chapter: Routes to N-Heterocyclic Carbene Complexes, Springer, Berlin, 2007 Search PubMed.
- (a) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 14180–14181 Search PubMed; (b) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 7052–7055 Search PubMed; (c) O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530–5533 Search PubMed.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer, 3rd, P. V. R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069–1071 Search PubMed.
- (a) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer, 3rd, P. V. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970–14971 Search PubMed; (b) M. Y. Abraham, Y. Wang, Y. Xie, P. Wei, H. F. Schaefer, 3rd, P. R. von Schleyer and G. H. Robinson, Chem.–Eur. J., 2010, 16, 432–435 Search PubMed.
- A. Sidiropoulos, C. Jones, A. Stasch, S. Klein and G. Frenking, Angew. Chem., Int. Ed., 2009, 48, 9701–9704 Search PubMed.
- (a) C. A. Dyker and G. Bertrand, Science, 2008, 321, 1050–1051 Search PubMed; (b) Y. Wang and G. H. Robinson, Chem. Commun., 2009, 5201–5213 Search PubMed; (c) Y. Wang and G. H. Robinson, Dalton Trans., 2012, 41, 337–345 Search PubMed; (d) Y. Wang and G. H. Robinson, Inorg. Chem., 2011, 50, 12326–12337 Search PubMed; (e) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 Search PubMed.
- (a) A. J. Arduengo III, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530–5534 Search PubMed; (b) A. J. Arduengo III, Acc. Chem. Res., 1999, 32, 913–921 Search PubMed.
- (a) A. Luque and S. Hegedus, Handbook of Photovoltaic Science and Engineering, Wiley, Chichester, 2003 Search PubMed; (b) B. G. Gribov and K. V. Zinov'ev, Inorg. Mater., 2003, 39, 653–662 Search PubMed.
- (a) C. J. Wilkins, J. Chem. Soc., 1953, 3409 Search PubMed; (b) R. Martens and W.-W. D. Mont, Chem. Ber., 1992, 125, 657–658 Search PubMed.
- (a) T. Böttcher, O. Shyshkov, M. Bremer, B. S. Bassil and G.-V. Röschenthaler, Organometallics, 2012, 31, 1278–1280 Search PubMed; (b) T. Böttcher, B. S. Bassil and G.-V. Röschenthaler, Inorg. Chem., 2012, 51, 763–765 Search PubMed.
- F. Meyer-Wegner, A. Nadj, M. Bolte, N. Auner, M. Wagner, M. C. Holthausen and H.-W. Lerner, Chem.–Eur. J., 2011, 17, 4715–4719 Search PubMed.
- (a) S. Nozakura and S. Konotsune, Bull. Chem. Soc. Jpn., 1956, 29, 322–326 Search PubMed; (b) R. A. Benkeser, Acc. Chem. Res., 1971, 4, 94–100 Search PubMed; (c) R. A. Benkeser and W. E. Smith, J. Am. Chem. Soc., 1968, 90, 5307–5309 Search PubMed; (d) W. W. duMont, L. P. Muller, L. Muller, S. Vollbrecht and A. Zanin, J. Organomet. Chem., 1996, 521, 417–419 Search PubMed.
- (a) R. S. Ghadwal, S. S. Sen, H. W. Roesky, G. Tavcar, S. Merkel and D. Stalke, Organometallics, 2009, 28, 6374–6377 Search PubMed; (b) N. Kuhn, T. Kratz, D. Bläser and R. Boese, Chem. Ber., 1995, 128, 245–250 Search PubMed; (c) R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683–5686 Search PubMed.
- G. Engelhardt, R. Radeglia, H. Jancke, E. Lippmaa and M. Mägi, Org. Magn. Reson., 1973, 5, 561–566 Search PubMed.
- D. Tapu, D. A. Dixon and C. Roe, Chem. Rev., 2009, 109, 3385–3407 Search PubMed.
- O. Hollóczki and Ls Nyulászi, Organometallics, 2009, 28, 4159–4164 Search PubMed.
- Z. Liu, J. Phys. Chem. A, 2009, 113, 6410–6414 Search PubMed.
- F. M. Bickelhaupt and E. J. Baerends, Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry, Wiley-VCH, Hoboken, 2000 Search PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- Ev. Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597 Search PubMed.
- (a) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 Search PubMed; (b) C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 Search PubMed.
- T. Stefan and R. Janoschek, J. Mol. Model., 2000, 6, 282–288 Search PubMed.
- R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 711–724 Search PubMed.
- (a) C. Boehme and G. Frenking, J. Am. Chem. Soc., 1996, 118, 2039–2046 Search PubMed; (b) C. Heinemann, T. Müller, Y. Apeloig and H. Schwarz, J. Am. Chem. Soc., 1996, 118, 2023–2038 Search PubMed.
- Y. Wang, Y. Xie, M. Y. Abraham, R. J. Gilliard, P. Wei, H. F. Schaefer, P. V. R. Schleyer and G. H. Robinson, Organometallics, 2010, 29, 4778–4780 Search PubMed.
- A. J. Arduengo III, F. Davidson, H. V. R. Dias, J. R. Goerlich, D. Khasnis, W. J. Marshall and T. K. Prakasha, J. Am. Chem. Soc., 1997, 119, 12742–12749 Search PubMed.
- (a) S. Khan, S. S. Sen and H. W. Roesky, Chem. Commun., 2012, 48, 2169–2179 Search PubMed; (b) J. Petuskova, M. Patil, S. Holle, C. W. Lehmann, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2011, 133, 20758–20760 Search PubMed.
- B. D. Ellis, C. A. Dyker, A. Decken and C. L. Macdonald, Chem. Commun., 2005, 1965–1967 Search PubMed.
- A. M. Voutchkova, L. N. Appelhans, A. R. Chianese and R. H. Crabtree, J. Am. Chem. Soc., 2005, 127, 17624–17625 Search PubMed.
- M. Azouri, J. Andrieu, M. Picquet, P. Richard, B. Hanquet and I. Tkatchenko, Eur. J. Inorg. Chem., 2007, 4877–4883 Search PubMed.
- (a) A. Schmidpeter, S. Lochschmidt and A. Willhalm, Angew. Chem., 2006, 95, 561–562 Search PubMed; (b) H. W. Wanzlick and E. Schikora, Angew. Chem., 1960, 72, 494 Search PubMed.
- K. B. Dillon, A. W. G. Platt, A. Schmidpeter, F. Zwaschka and W. S. Sheldrick, Z. Anorg. Allg. Chem., 1982, 488, 7–26 Search PubMed.
- (a) J. J. Weigand, K. O. Feldmann and F. D. Henne, J. Am. Chem. Soc., 2010, 132, 16321–16323 Search PubMed; (b) A. C. Filippou, O. Chernov and G. Schnakenburg, Angew. Chem., Int. Ed., 2009, 48, 5687–5690 Search PubMed.
- M. B. Giudicelli, D. Picq and B. Veyron, Tetrahedron Lett., 1990, 31, 6527–6530 Search PubMed.
- M. F. Lappert and P. L. Pye, J. Chem. Soc., Dalton Trans., 1977, 2172 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, crystallographic data, and thermogravimetric analysis. CCDC reference numbers 895219=895225. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21214e |
| This journal is © The Royal Society of Chemistry 2013 |