Bioorthogonal reactions challenged: DNA templated native chemical ligation during PCR†
Alexander
Roloff
and
Oliver
Seitz
*
Institut für Chemie der Humboldt-Universität zu Berlin, Brook-Taylor-Strasse 2, 12489-Berlin, Germany. E-mail: oliver.seitz@chemie.hu-berlin.de; Fax: +49 30-2093-7266; Tel: +49 30-2093-7446
First published on 3rd October 2012
Abstract
DNA directed chemistry is commonly performed by using nanomolar amounts of DNA templates. Herein we introduce a method that allows the use of attomolar template loads. A DNA templated native chemical ligation yields to the covalent fixation of two fluorophores while the template is being produced during polymerase chain reaction.
Introduction
DNA directed synthesis offers fascinating opportunities for the programmable formation of functional molecules. For example, DNA templated chemistry has been used to facilitate drug screening.1 In the pursuit of so-called molecular doctors2 nucleic acid templates served to encode the formation of cell toxic singlet oxygen3 or proapoptotic peptides.4 Other applications were aimed at the synthesis of functional materials such as superconducting nanowires5 or arbitrarily shaped metal nanoparticles,6 as well as signaling molecules for diagnostic purposes.7 Among the various DNA-templated reactions available only a few have been demonstrated to proceed in complex biomolecular environments. Notable examples are DNA/RNA templated Staudinger reactions8 and phosphorothioate based SN2 reactions.9Though nucleic acid directed reactions can enable turnover in template,10 most chemistries typically require rather high concentrations of the template nucleic acids. To overcome this restriction it would be desirable to link these templated reactions with enzymatic amplification of the template. Very recently it has been shown by Kool that templated reactions can indeed proceed during isothermal rolling circle amplification (RCA) of the target sequence.11 However, RCA is a linear process resulting in rather low amplification rates, so that the reaction at low picomolar template concentrations was inefficient. On the contrary, the polymerase chain reaction (PCR) is the most frequently applied method to exponentially amplify minute DNA amounts. If PCR were interfaced with DNA templated chemistry, extremely low amounts of DNA would suffice to instruct the formation of functional molecules while maintaining the extraordinary sequence specificities12 provided by DNA templated reactions.
According to this strategy, bioorthogonal DNA templated chemistry needs to be challenged with the extreme conditions during PCR. Chemoselectivity has to be maintained in the presence of proteins and nucleic acids at temperatures up to 95 °C without increasing off-template reactions and/or affecting the PCR efficiency. In this study we show that a DNA templated chemical reaction can be performed while the template is being produced in PCR.
In earlier studies we equipped peptide nucleic acids (PNA)13 with thioester and 1,2-aminothiol moieties required to drive DNA templated native chemical ligation based reactions.12 PNA has a very high affinity for complementary DNA and RNA and shows excellent sequence specificity.14 Thus, concentrations of the PNA oligomers can be kept low, thereby minimizing the reaction in absence of the template DNA. We showed that DNA directed reactions proceeded on long DNA templates produced by PCR.12b,15 However, the reaction during PCR was not attempted because at the elevated temperatures applied during thermal cycling the template controlled ligation was outcompeted by background ligation and the hydrolysis of the used PNA thioesters.
Results and discussion
We set out to develop a DNA triggered reaction that proceeds while the template is being produced by a polymerase chain reaction (Scheme 1). Given the constraints imposed by the PCR process several requirements have to be fulfilled. The reaction must tolerate the high temperature required in the denaturation step of PCR (step 1 in Scheme 1). Furthermore, the reactive PNA conjugates must be designed to allow binding of the template during primer annealing (step 2 in Scheme 1), which will lead to the formation of the quaternary complex 1. This will promote a subsequent proximity enhanced ligation. Importantly, the complex 2 comprised of the DNA template and the formed ligation product must not be too stable in order to allow the DNA polymerase to read through the template during primer extension (step 3 in Scheme 1). To enable real-time monitoring of the ligation reaction, the reactive molecules were equipped with 6-carboxyfluorescein (FAM) and 6-carboxytetramethylrhodamine (TMR) dyes. Product formation will be accompanied by a decrease in FAM emission and an increase of TMR emission due to fluorescence resonance energy transfer (FRET).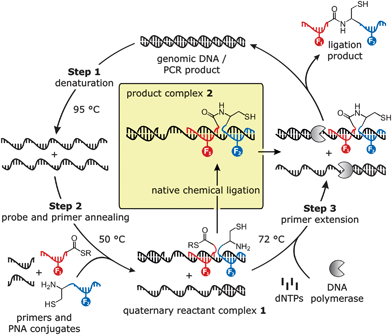 | ||
| Scheme 1 DNA templated native chemical ligation during PCR (F1 = FAM, F2 = TMR). | ||
We tested native chemical PNA ligation on a short single stranded DNA template harboring the somatic V600E (T1799A) point mutation on the human BRaf-gene, which occurs at high frequency in human malignant melanoma (Fig. 1a).16 The PNA oligomers had to be of sufficient length to assure hybridization with the template during the primer annealing step of PCR. This excluded fluorophore attachment to their terminal ends, as the donor–acceptor distance in the ligation product would be too large for an efficient FRET. In consequence, internal labeling on the PNA-backbone was accomplished by using lysine-derived PNA monomers (see ESI†).17 At the outset of our work, a solution to the problem of background ligation and thioester hydrolysis at high temperatures was sought. Native chemical ligation reactions are usually performed by using α-amino acid thioesters such as the glycine thioester in 3. We assumed that the introduction of an additional methylene group in the β-alanine thioester 4 lowers the off-template reaction rates by decreasing the electrophilicity of the thioester moiety. The FAM-labeled PNA-Gly-thioester 3 and the PNA-βAla-thioester 4 were exposed to conditions that would facilitate hydrolysis. At pH 8.5 and 50 °C about 50% of the Gly-thioester was hydrolyzed after 2 h, whereas more than 60% of the βAla-thioester remained stable even after 8 h under the same conditions (Fig. 1b, also see Fig. S10†).
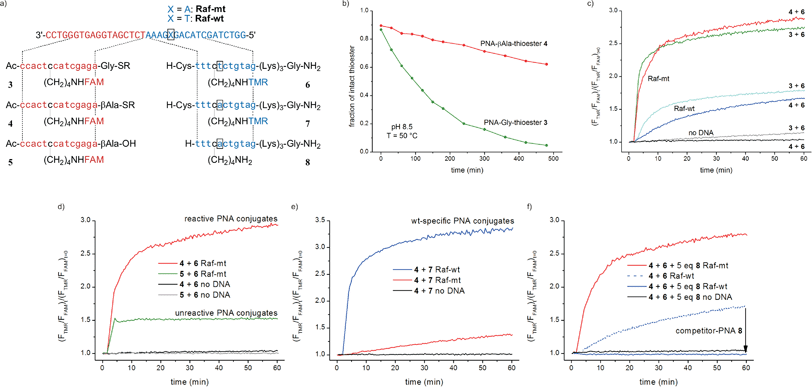 | ||
| Fig. 1 (a) DNA templates and PNA conjugates. (b) Hydrolysis of PNA-Gly-thioester 3 and PNA-βAla-thioester 4 at pH 8.5 and 50 °C (2 μM thioester, 100 mM NaH2PO4). Relative increase of FRET signal during (c) reaction of 3 or 4 with mt-specific TMR-labeled Cys-PNA 6 compared with (d) adjacent hybridization of unreactive pair 5 + 6; or the reaction of (e) wt-specific pair 4 + 7 and f) 4 + 6 in presence of 5 eq. competitor-PNA 8 (λex. = 470 nm, λem. = 523 nm (FAM), 585 nm (TMR), 1 μM PNA conjugates, 1 μM DNA template when added, 10 mM NaH2PO4, 150 mM NaCl, 10 mM MESNA, pH 7.4, 50 °C, R = (CH2)2SO3H). | ||
Next, mutually reactive pairs (TMR-labeled Cys-PNA 6 with either Gly-thioester 3 or βAla-thioester 4) were allowed to react at 1 μM concentration and 50 °C in both the presence and absence of the complementary DNA-template (Fig. 1c). Surprisingly, though βAla-thioester 4 showed a reduced reactivity in hydrolysis and background reactions (see no template reactions in Fig. 1c), when 1 eq. of matched template Raf-mt was added, both reactions were accompanied by a similar 2.8 fold increase in the FRET signal and showed a comparable kinetic profile. The templated reactions proceeded rapidly, as required for reactions that ought to be performed during the short time available during PCR. Within 15 minutes about 80% of the maximum signal increase was obtained. Of note, the 4 + 6 pair allowed for ca. 5-fold higher rate acceleration (971) than the 3 + 6 pair (216, see Fig. S11†). Furthermore, reactions on single base mismatched DNA Raf-wt revealed that the β-alanine thioester in 4 yielded a more sequence specific signal than the glycine thioester in 3 during reaction with Cys-PNA 6.
The remarkable stability against hydrolysis, the decreased background ligation at elevated temperatures, the high rate of the templated reaction and the higher sequence specificity recommended βAla-thioester 4 for further evaluation. However, the observed changes of the fluorescence ratios do not necessarily reflect PNA ligation, but could also arise from adjacent hybridization. We deliberately hydrolyzed thioester 4 and obtained the free carboxylic acid 5. The signal gained upon addition of template to the unreactive pair 5 + 6 evolved instantaneously, probably because hybridization proceeds within milliseconds, and remained constant over time (Fig. 1d). On the contrary, the reactive oligomers 4 + 6 yielded a significantly higher signal which increased over time and asymptotically reached a plateau. This comparison demonstrates the advantage of chemical fixation of donor and acceptor-dye in the product duplex. The different behavior is due to the high temperature and the differences between measurements of equilibrium and non-equilibrium systems. The TM of duplex 4·Raf-mt was 67 °C compared to 47 °C for duplex 6·Raf-mt. At temperatures near the TM only half of PNA 6 will be annealed and this fraction remains constant over time. By contrast, the reactive PNA set leads to the formation of a dually labeled product; the majority of which will be bound to the template. The fraction of unbound template will continue to trigger the formation of new product molecules until the concentration of free template or reactive molecules is too low to support annealing.
We next studied the reaction of control PNA conjugates that would furnish positive signals upon reaction on wildtype templates. Expectedly, the reaction between PNA thioester 4 and wt-specific Cys-PNA 7 proceeded much faster on the matched template Raf-wt than on Raf-mt (Fig. 1e). The 68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 match/mismatch selectivity (see Fig. S13†) was significantly higher than the selectivity provided by the mutant specific PNA 6 (14:1). To improve the specificity of the mutant specific ligation, we included an unlabeled, non-thiolated “competitor-PNA” 8 which is complementary to the wildtype-target and competes with the reactive, mt-specific Cys-PNA 6 for template binding. In the presence of Raf-wt, the ternary complex will be 4·8·Raf-wt (favored over 4·6·Raf-wt) and consequently chemical ligation of 4 with 6 would be repressed. Since 8 has a single base mismatch with Raf-mt, there should be little harm to product formation on this template. Indeed, even a fivefold excess of the competitor PNA 8 (5 μM) had only marginal effects on the signal increase in the presence of Raf-mt, whereas the mismatch-templated reaction was completely suppressed (Fig. 1f). Noteworthy, the signal increase was even lower than for the template independent background reaction (see Fig. S14†). This can be explained by the coexistence of ternary complex 4·8·Raf-wt and unbound Cys-PNA 6: neither in the hybridized nor in the unbound state is the concentration of both reactive PNA molecules high enough for a successful ligation event.
:1 match/mismatch selectivity (see Fig. S13†) was significantly higher than the selectivity provided by the mutant specific PNA 6 (14:1). To improve the specificity of the mutant specific ligation, we included an unlabeled, non-thiolated “competitor-PNA” 8 which is complementary to the wildtype-target and competes with the reactive, mt-specific Cys-PNA 6 for template binding. In the presence of Raf-wt, the ternary complex will be 4·8·Raf-wt (favored over 4·6·Raf-wt) and consequently chemical ligation of 4 with 6 would be repressed. Since 8 has a single base mismatch with Raf-mt, there should be little harm to product formation on this template. Indeed, even a fivefold excess of the competitor PNA 8 (5 μM) had only marginal effects on the signal increase in the presence of Raf-mt, whereas the mismatch-templated reaction was completely suppressed (Fig. 1f). Noteworthy, the signal increase was even lower than for the template independent background reaction (see Fig. S14†). This can be explained by the coexistence of ternary complex 4·8·Raf-wt and unbound Cys-PNA 6: neither in the hybridized nor in the unbound state is the concentration of both reactive PNA molecules high enough for a successful ligation event.
We explored native chemical PNA ligation in real-time PCR. First, we evaluated whether the PCR process tolerates the presence of thiol additives commonly added to help maintain a reducing environment in native chemical ligation. Wildtype human genomic DNA (WT-HGD) was selected as the template for PCR, and a primer set amplifying a region from exon 15 of the BRaf-gene containing the T1799A point mutation site was used (for target and primer sequences see Fig. S15†). Conventional SYBR-Gold monitoring and subsequent PAGE-analysis revealed that 10 mM sodium 2-mercaptoethanesulfonate (MESNA) is well tolerated (see Fig. S16†). To exclude that the PNA conjugates inhibit amplification, we conducted a PCR with variable concentrations of FAM-labeled PNA-thioester 4 and TMR-labeled Cys-PNA 7. The PCR proceeded efficiently despite the presence of up to 1 μM PNA oligomers as judged from PAGE analysis (see Fig. S17†). Next, the reaction between 4 and 7 during PCR was monitored (Fig. 2a). During each cycle in the 30 s primer-annealing step the FAM fluorophore was excited and FAM- and TMR-emissions were recorded at 530 and 585 nm, respectively. However, after an initial increase in the FRET signal after 23 cycles, a plateau at low signal intensity was reached after 30 cycles. We attributed this to a limited access of probes to the target strand, as template reannealing competes with probe hybridization especially in later cycles of PCR. This was remedied by applying asymmetric PCR conditions (50 nM reverse primer, 400 nM forward primer), which provide an excess of single stranded PCR product. Under these conditions the signal continued to evolve even at later cycle numbers. Experiments with the unreactive PNA set 5 + 7 confirmed that this signal increase was due to the reaction and not adjacent hybridization (Fig. S18†). It should be stated that longer FRET-based hybridization probes function very well in the sequence specific detection of PCR products.18 However, the chemical ligation of two shorter probes provides options for increasing the sequence selectivity and for permanently joining two functional entities in a DNA-dependent manner beyond FRET-based signaling.
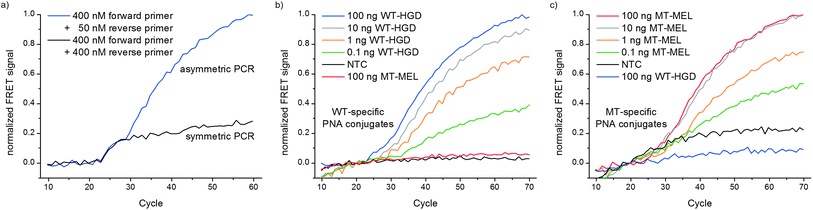 | ||
| Fig. 2 Normalized FRET signal upon PNA ligation during PCR. (a) Symmetric PCR (400 nM each primer) or asymmetric PCR (400 nM forward primer, 50 nM reverse primer), FAM-labeled PNA–thioester 4 and TMR-labeled WT-specific Cys-PNA 7 (200 nM PNA conjugates, 100 ng template WT-HGD); (b) 4 and WT-specific Cys-PNA 7 or (c) 4 and MT-specific Cys-PNA 6 in presence of 100–0.1 ng match DNA, 100 ng mismatch DNA and without DNA (PCR conditions: 400 nM forward primer, 50 nM reverse primer, 200 μM dNTPs, 2.5 mM MgCl2, 300 nM PNA conjugates, 1 mM MESNA, 10 mM TRIS, pH 8.5 (at 25 °C), 1 u Taq-Pol, PCR protocol: 10 s. 95 °C, 30 s. 50 °C (detection), 20 s. 72 °C, λex. = 490 nm (FAM), 485 nm (TMR), λem. = 530 nm (FAM), 585 nm (TMR)). | ||
We tested the scope of the “in PCR ligation” by reducing the starting amount of template and also investigated the sequence specificity by using genomic DNA from the human melanoma cell line SK-MEL28 as mutant template (MT-MEL), known to harbor the T1799A point mutation.16a Hence, the PCR mix contained either the WT-specific pair 4 + 7 or the MT-specific pair 4 + 6 as well as the templates WT-HGD or MT-MEL. As little as 0.1 ng template corresponding to approximately 30 template molecules (in 20 μL reaction volume this corresponds to a concentration of ca. 2.5 aM) could be distinguished from the no template control (NTC). These results demonstrate that attomolar concentrations of human genomic DNA are sufficient to trigger the formation of a functional molecule. The FRET signal emerged only in the presence of the fully matched templates for both combinations, whereas the single base mismatched DNAs were indistinguishable from the NTC (Fig. 2b and c). PAGE-analysis of the PCR products verified that the expected amplicons were produced in all cases (Fig. S19 and S20†). In quantitative PCR measurements threshold cycle values are usually used to determine starting amounts of templates. However, as the presented system is based on asymmetric amplification and includes a chemical reaction that is principally capable of turnover in template (especially for lower template loadings), this is not appropriate in this case. Nevertheless it can be estimated that after 30 to 40 (WT-specific PNA conjugates) or 35 to 45 (MT-specific PNA conjugates) cycles, a reliably detectable signal emerges depending on the starting concentration of the template.
Conclusions
In this study we presented a bioorthogonal DNA templated chemical reaction that is conducted during a PCR and directly triggered by the amplified DNA. Our reaction system tolerates a wide range of temperatures up to 95 °C and allows real-time detection of product formation. The DNA templated synthesis is enabled starting from attomolar concentrations of biologically originated templates with single nucleotide specificity. The interfacing with PCR may provide a general solution to the problem of product inhibition, which limits DNA-templated ligation reactions.10 The high degree of chemoselectivity recommends templated native chemical ligation for other applications. For example, it may prove feasible to generate bi- or even multivalent ligands by sequence programmed synthesis or to diversify DNA-libraries in SELEX (systematic evolution of ligands by exponential enrichment) procedures. The template directed chemistry may proceed within cells, enabling for instance real-time detection of mRNA expression levels. The advantages of PNA-based probes in live cell mRNA imaging have recently been reported.19 In ongoing work we investigate whether PNA-based reaction systems yielding products with low template affinities can make enzymatic template amplifications redundant.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft.Notes and references
- For examples see: (a) Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601 CrossRef CAS; (b) M. H. Hansen, P. Blakskjær, L. K. Petersen, T. H. Hansen, J. W. Højfeldt, K. V. Gothelf and N. J. V. Hansen, J. Am. Chem. Soc., 2009, 131, 1322 CrossRef CAS; (c) G. Georghiou, R. E. Kleiner, M. Pulkoski-Gross, D. R. Liu and M. A. Seeliger, Nat. Chem. Biol., 2012, 8, 366 CrossRef CAS ; for a review see: R. E. Kleiner, C. E. Dumelin and D. R. Liu, Chem. Soc. Rev., 2011, 40, 5707 Search PubMed.
- M. F. Jacobsen, E. Cló, A. Mokhir and K. V. Gothelf, ChemMedChem, 2007, 2, 793 CrossRef CAS ; and references cited therein.
- D. Arian, E. Cló, K. V. Gothelf and A. Mokhir, Chem.–Eur. J., 2010, 16, 288 CrossRef CAS.
- A. Erben, T. N. Grossmann and O. Seitz, Angew. Chem., 2011, 123, 2880 ( Angew. Chem., Int. Ed. , 2011 , 50 , 2828 ) CrossRef CAS; A. Erben, T. N. Grossmann and O. Seitz, Bioorg. Med. Chem. Lett., 2011, 21, 4993 CrossRef.
- D. S. Hopkins, D. Pekker, P. M. Goldbart and A. Bezryadin, Science, 2005, 308, 1762 CrossRef CAS.
- R. Schreiber, S. Kempter, S. Holler, V. Schüller, D. Schiffels, S. S. Simmel, P. C. Nickels and T. Liedl, Small, 2011, 7, 1795 CrossRef CAS.
- For a recent review see: A. Shibata, H. Abe and Y. Ito, Molecules, 2012, 17, 2446 CrossRef CAS.
- (a) Z. Pianowski, K. Gorska, L. Oswald, C. A. Merten and N. Winssinger, J. Am. Chem. Soc., 2009, 131, 6492 CrossRef CAS; (b) R. M. Franzini and E. T. Kool, J. Am. Chem. Soc., 2009, 131, 16021 CrossRef CAS; (c) K. Gorska, I. Keklikoglou, U. Tschulena and N. Winssinger, Chem. Sci., 2011, 2, 1969 RSC.
- (a) H. Abe and E. T. Kool, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 263 CrossRef CAS; (b) A. P. Silverman, E. J. Baron and E. T. Kool, ChemBioChem, 2006, 7, 1890 CrossRef CAS.
- (a) T. N. Grossmann, A. Strohbach and O. Seitz, ChemBioChem, 2008, 9, 2185 CrossRef CAS; (b) H. Abe and E. T. Kool, J. Am. Chem. Soc., 2004, 126, 13980 CrossRef CAS; (c) T. N. Grossmann and O. Seitz, J. Am. Chem. Soc., 2006, 128, 15596 CrossRef CAS; (d) T. N. Grossmann and O. Seitz, Chem.–Eur. J., 2009, 15, 6723 CrossRef CAS.
- E. M. Harcourt and E. T. Kool, Nucleic Acids Res., 2012, 40, e65 CrossRef CAS.
- (a) A. Mattes and O. Seitz, Chem. Commun., 2001, 2050 RSC; (b) S. Ficht, A. Mattes and O. Seitz, J. Am. Chem. Soc., 2004, 126, 9970 CrossRef CAS; (c) S. Ficht, C. Dose and O. Seitz, ChemBioChem, 2005, 6, 2098 CrossRef CAS.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497 CAS.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566 CrossRef CAS.
- For a templated Wittig reaction on DNA from PCR see: X.-H. Chen, A. Roloff and O. Seitz, Angew. Chem., 2012, 124, 4556 ( Angew. Chem., Int. Ed. , 2012 , 51 , 4479 ) CrossRef.
- (a) H. Davies, et al. , Nature, 2002, 417, 949 CrossRef CAS; (b) P. M. Pollock, et al. , Nat. Genet., 2003, 33, 19 CrossRef CAS , (see ESI† for complete author list).
- (a) C. Dose, S. Ficht and O. Seitz, Angew. Chem., 2006, 118, 5495 ( Angew. Chem., Int. Ed. , 2006 , 45 , 5369 ) CrossRef; (b) C. Dose and O. Seitz, Bioorg. Med. Chem., 2008, 16, 65 CrossRef CAS.
- (a) C. T. Wittwer, K. M. Ririe, R. V. Andrew, D. A. David, R. A. Gundry and U. J. Balis, BioTechniques, 1997, 22, 176 CAS; (b) P. S. Bernard, G. H. Pritham and C. T. Wittwer, Anal. Biochem., 1999, 273, 221 CrossRef CAS.
- (a) S. Kummer, A. Knoll, E. Socher, L. Bethge, A. Herrmann and O. Seitz, Angew. Chem., 2011, 123, 1972 ( Angew. Chem., Int. Ed. , 2011 , 50 , 1931 ) CrossRef; (b) S. Kummer, A. Knoll, E. Socher, L. Bethge, A. Herrmann and O. Seitz, Bioconjugate Chem., 2012 DOI:10.1021/bc300249f.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of modified PNA monomers and fluorophore labeled PNA conjugates as well as experimental details are described in the ESI. See DOI: 10.1039/c2sc20961f |
| This journal is © The Royal Society of Chemistry 2013 |