Switchable anionic surfactants for the remediation of oil-contaminated sand by soil washing†
Elize Ceschia,
Jitendra R. Harjani,
Chen Liang,
Zahra Ghoshouni,
Tamer Andrea,
R. Stephen Brown* and
Philip G. Jessop*
Department of Chemistry, Queen's University, 90 Bader Lane, Kingston, ON K7L 3N6, Canada. E-mail: jessop@chem.queensu.ca; Fax: +1 (613) 533-6669; Tel: +1 (613) 533-3212
First published on 3rd December 2013
Abstract
Soil remediation requires technologies to restore contaminated soil to a state that is environmentally acceptable. In most cases, while the soil can be remediated the contaminant itself cannot be reclaimed. However, ex situ soil washing with an aqueous solution of a surfactant would allow easy recovery of the contaminant if the resulting emulsion could be easily and reliably broken. CO2-responsive switchable surfactants that can be switched on and off by CO2 addition and removal would allow facile emulsion breaking, but all reported examples are cationic surfactants. Because soil washing requires anionic surfactants, we have investigated phenolate, benzoate and carboxylate salts as switchable anionic surfactants. These surfactants can indeed be switched on and off with CO2. Three switchable anionic surfactants (sodium 2-nitro-4-((octyloxy)carbonyl)phenolate (1a), sodium 4-(octyloxy) benzoate (3a) and sodium laurate (4a)) and two commercial nonswitchable surfactants (sodium dodecyl sulfate (SDS) and Triton X-100) were evaluated for their ability to wash Ottawa Sand artificially contaminated with North Sea crude oil. At 50 °C, all three switchable surfactants were able to remove North Sea crude oil from sand and have the added feature of facile oil separation and recovery from the wash mixture after CO2 treatment.
1 Introduction
Ex situ soil washing is one of the most versatile contaminated soil remediation techniques because it can be applied to virtually all target organic contaminants: e.g. petroleum hydrocarbons, metals, PCBs, pesticides and PAHs.1 The goals of soil washing are to remove the contaminant from the soil and to concentrate the contaminants for further treatment.2 Normally, the soil is first screened and divided into two fractions: sand and fines (silt and clay). Sand, the subject of the present study, is the major component and is easier to treat because of the larger particle size and lower organic matter content.3 After size separation, any volatile contaminants present in the fractions are removed by heating the soil. Soil washing is usually performed with hot aqueous solutions of surfactants because higher temperatures help to reduce the viscosity of contaminants like oil4 and increase the solubilizing ability of the surfactants, although the higher temperature increases energy costs. The cleaned soil is then removed and the wash water is treated and recycled. Oil-containing sludge, contaminated fines, and contaminated water are sent to wastewater treatment facilities.In soil washing, surfactants are used to liberate the oil from the soil in part by reducing the interfacial tension between the oil and the water, as well as decreasing the attraction between the contaminant and the soil. However, surfactants tend to continue to stabilize an emulsion of the contaminant oil in water long after the washing is performed, making it difficult to separate the oil from the water. Switchable surfactants, which can switch off upon command, could facilitate both the washing and then the subsequent oil/water separation.
Switchable surfactants have surface activity that can be switched “on” or “off” upon application of a trigger such as light,5 pH6,7 or redox reagents.8–16 The switchable cationic surfactants developed by Jessop and co-workers are pH switchable and designed specifically to respond to pH changes induced by the introduction and removal of CO2 from solution.17,18 To date, these switchable surfactants have been found to have the ability to stabilize oil/water emulsions,17,18 and to have potential utility in emulsion polymerization19,20 and the transport of very heavy crude oils through pipelines.21
Switchable surfactants triggered by CO2 seemed ideal for a process for soil washing because they could facilitate oil and surfactant recovery, but the reported examples of such surfactants are all cationic. Cationic surfactants do not perform well in soil washing because they bind to anionic sites in the solid material.22 Not surprisingly, switchable cationic surfactants are also ineffective for soil washing. We therefore needed to find CO2-triggered switchable anionic surfactants (SAS).
A SAS would be expected to function as an emulsion-stabilizing surfactant in the absence of CO2, while the SAS is in its anionic form (A− in eqn (1)). Addition of CO2 would convert it into its neutral form (HA), which should not be able to stabilize emulsions. Removal of CO2 should reverse the reaction. One would expect this transformation to be possible provided that the addition of CO2 lowers the pH well below the midpoint of the acid HA (i.e. the pH at which 50% of the SAS in the system is in the neutral form) and the removal of the CO2 restores the pH to a value well above the midpoint.
 | (1) |
Literature reports support the expectation that sodium carboxylate salts should be reversibly switchable. It has been known for many years that exposure of aqueous solutions of fatty acid sodium salts to CO2 causes the air/water surface tension to change.23 Moore and Lefevre24 described an emulsion polymerization using sodium oleate as the surfactant and styrene and acrylonitrile as the comonomers. After the latex was produced, they showed that it could be destabilized by the addition of CO2, presumably because the acid form of the surfactant was less effective than the anionic form at latex stabilization. The use of pressurized CO2 to lower the pH below 3.7, in order to break emulsions containing anionic surfactants, has also been described.25 Therefore, sodium oleate and related anionic surfactants appear to be switchable anionic surfactants. However, useful SAS would ideally require neither pressurized CO2 nor pH values as low as 3.7 to be switched off.
We propose that SAS could be used to facilitate the oil/water separation stage in the soil washing process, as shown in Fig. 1. After the washing is completed and the soil has been separated, the washing solution would be brought into a separator. CO2 would be introduced, causing the surfactant to be “turned off” and resulting in the separation of the crude oil and surfactant from the wash water. The crude can then be reclaimed and recycled, while the wash water would be decarbonated, recombined with the surfactant, and re-introduced into the washer.
 | ||
| Fig. 1 Proposed scheme for soil washing using switchable surfactants. For simplicity, stages such as soil screening, washing of oversize material, and separation of fines from the water are not shown. | ||
2 Results and discussion
2.1 CO2-Triggered switchable anionic surfactants
In our study of potential switchable anionic surfactants, we evaluated two phenolate sodium salts, one benzoate and one carboxylate (Scheme 1). | ||
| Scheme 1 Anionic surfactants tested for their response to CO2 addition and removal. In each case, “a” refers to the sodium salt (sodium phenolate or carboxylate) and “b” refers to the acid form (phenol or carboxylic acid). | ||
Switching the sodium phenolate surfactants (1a, 2a) off with CO2 and on again by removal of CO2 was tested by visual observation, 1H NMR spectroscopy, pH monitoring, and surface tension measurements. Clear yellow solutions of 1a were prepared in situ by dissolving 1b (5 mM) and NaOH (7.5 mM) in H2O (for the NMR experiments, D2O and 8 equivalents of Na2CO3 were used instead). The use of excess NaOH produced a pH of 11.4. After CO2 was introduced into the vial, the pH dropped to 5.6 (Table 1), phenol 1b precipitated, and the solution almost completely decolorized within 5 min (Fig. S1†), due to the poor solubility of the neutral phenol 1b. 1H NMR spectroscopy of the liquid phase showed that there was no longer any detectable quantity of either 1a or 1b in solution. Heating the mixture to 70 °C for 1 h to remove most of the CO2 raised the pH to 8.2, re-dissolved the precipitate and restored the yellow colour to the solution. NMR spectroscopy confirmed that the amount of dissolved 1a had returned to the original amount. Repeated cycles gave the same results, showing that the behaviour is fully and repeatedly reversible. This reversible change was also evident in the air/water surface tension of the aqueous solution, which was 57–63 dynes cm−2 in the presence of CO2 and 33–35 dynes cm−2 in the absence of CO2. Similar switching behaviour was observed with a solution of 2.
Introduction of CO2 into aqueous solutions of sodium carboxylate surfactants (3a, 4a) converts the carboxylate anion into the poorly soluble carboxylic acid; a 5 mM solution of sodium dodecanoate (4a) had a pH of 8.4, but after CO2 was bubbled through the solution for 10 min the pH dropped to 5.6 and dodecanoic acid precipitated as a white powder (Table 1). In order to switch the surfactant back to the anionic form, the solution was heated to 60 °C for 1 h, causing the CO2 to dissipate and the acid to redissolve as the pH rose to 8.2. Note that the switching of compounds 1a–4a from anionic form to the acid form occurs even though the pH never drops below the pKa of the acid form (see Section 2.2 for an explanation).
| Treatment | 1a [A−]0 = 5 mM | 2b [A−]0 = 5 mM | 3b [A−]0 = 5 mM | 4c [A−]0 = 5 mM |
|---|---|---|---|---|
| a pH of a solution of 1b (5 mM) and NaOH (7.5 mM) in H2O treated to alternating treatments of CO2 bubbling for 10 min followed by heating to 70 °C for 1 h.b pH of a solution of 2b (5 mM) or 3b (5 mM) and NaOH (7.5 mM) in H2O treated to alternating treatments of CO2 bubbling for 10 min followed by heating to 70 °C for 3 h.c pH of 5 mM sodium laurate (4a) solutions in H2O treated to alternating treatments of CO2 bubbling for 10 min followed by heating to 60 °C for 1 h.d pKa for the analogous ethyl ester.27e pKa for the analogous methyl ester, this work.f pKa for the analogous 4-butoxybenzoic acid.28 | ||||
| None | 11.4 | 11.7 | 11.2 | 8.4 |
| CO2 | 5.6 | 5.8 | 5.8 | 5.6 |
| 1 cycle | 8.2 | 9.5 | 8.6 | 8.2 |
| 1 cycle + CO2 | 5.6 | 5.8 | 5.9 | 5.8 |
| 2 cycles | 8.5 | 9.2 | 8.6 | 7.8 |
| 2 cycles + CO2 | 5.7 | 5.8 | 6.0 | 5.7 |
| 3 cycles | 8.3 | 9.0 | 8.4 | 8.1 |
| 3 cycles + CO2 | 5.8 | 5.9 | 5.9 | 5.6 |
| 4 cycles | 8.5 | 9.3 | 8.6 | 8.4 |
| 4 cycles + CO2 | 5.8 | 5.8 | 5.9 | 6.1 |
| 5 cycles | 8.7 | 9.1 | 8.4 | 8.9 |
| pKa of acid | 5.4d | 5.3e | 4.5f | 5.3 (ref. 26) |
Peters29 and Wakelin30 reported that the interfacial tension between benzene and aqueous glycine buffer solutions, in the presence of decanoic acid (58 mM relative to water content), is roughly constant at ∼30 dynes cm−1 (cf. 35 with benzene/water alone) at pH values below 7 but drops dramatically if the pH is raised to 8 or 9; the value at pH 8.2 is 24 dynes cm−1. Decanoate is much more effective than decanoic acid at lowering the benzene/water interfacial tension. This justifies referring to the acid form of alkanoic acids as “switched off” surfactants relative to the sodium alkanoate salts.
We found that emulsions of 1-octanol and water, stabilized with sodium dodecanoate, were stable for hours if prepared under air but separated immediately if prepared in a CO2-saturated environment (Fig. S3†). The phenolate surfactants, like the carboxylates, are capable of stabilizing 1-octanol/water emulsions in the absence of CO2 but not in its presence (Fig. S2†).
2.2 The pH values required for switching SAS
Switching of an anionic surfactant A− between its anionic form and its neutral form (eqn (3)) requires using CO2 to adjust the pH back and forth between two values, one above and one below the midpoint. For these purposes, the midpoint is defined as the pH at which half of the surfactant in the system (not merely that in the aqueous phase) has been protonated to the neutral form HA. We can predict the pH of the midpoint, and therefore the pH values required for switching, by using a simple mathematical model of the solubility and reaction equilibria.Considering an aqueous solution of A−, at an initial concentration of [A−]0 and in the absence of an oil phase, the pH required to “switch off” an anionic surfactant A− (eqn (3)) depends on the pKa of the neutral form HA, the solubility of HA, and the initial concentration of A− (i.e. [A−]0). In the unlikely situation that both A− and HA are completely soluble at all relevant concentrations, then the % protonation of A− is only dependent on the pH and the pKa (eqn 2 and 3) and the midpoint occurs when the pH matches the pKa. In this scenario, the CO2 treatment would have to reduce the pH to a value significantly below the pKa in order to effectively switch off the surfactant. Penth25 recommended a pH of 3.3 in order to break emulsions stabilized by carboxylate surfactants. In our experiments, we did not achieve a pH that low and yet we were still able to switch off the surfactant.
| A−(aq) + H3O+(aq) ⇌ HA(aq) + H2O(l) 1/Ka,HA | (2) |
 | (3) |
If the neutral form of the carboxylic acid has a solubility limit below 0.5[A−]0, then the pH of the midpoint will be shifted to a value higher than the pKa. This is often the case for carboxylate soaps because the critical micelle concentration (CMC) of NaA (23–28 mM for sodium dodecanoate)31,32 is usually much higher than the solubility limit for the neutral acid. Thus the switching of the anionic surfactants can be achieved by using CO2 to adjust the pH between two values that straddle the midpoint and that are both well above the pKa.
The pH-dependent solubility and the pH-dependence of the % protonation can be modelled mathematically by assuming that the sodium salt NaA is soluble at all relevant concentrations while HA has an intrinsic solubility limit S0. While S0 is pH-independent, the observed solubility S (the solubility of HA and A− combined) is pH-dependent and follows the Henderson–Hasselbalch equation (eqn (4)).33 The intrinsic solubility of protonated surfactants is rarely published, but it can be estimated from the observed solubility of the acid in pure water (Saq, eqn (5)).34 This method is not particularly accurate because it relies on a single data point, but it usefully generates an approximate value of S0. Fortunately, the S0 of dodecanoic acid was reported by Nyren and Back to be 2.3 × 10−5 M.26 The pH-dependent solubility, calculated using eqn (5), is shown for dodecanoate/dodecanoic acid in Fig. 2; this graph is identical to the one reported by Nyren and Back. As the pH is lowered by the addition of acid or CO2, the pH at which the surfactant solubility drops dramatically is well above the pKa.
| S = S0 (1 + Ka/[H3O+]) | (4) |
 | (5) |
 | ||
| Fig. 2 The pH-dependent solubility of dodecanoic acid (solid line) in water calculated using eqn (4) and an S0 value of 2.3 × 10−5 M. Also shown is the pH dependence of the % protonation of a 5 mM (nominal concentration) dodecanoate solution calculated using eqn (3) and (6) assuming that dodecanoic acid in its neutral form has (a) infinite solubility (dotted line) or (b) a solubility limit of 2.3 × 10−5 M (dashed line). The midpoints are at pH 5.3 and 7.3, respectively. | ||
The pH dependence of the % protonation of A− is governed by eqn (4) for conditions in which [HA] < S0 and by eqn (6) for conditions in which [HA] = S0. This calculates the % protonation for all of the A−/HA in the system, not just that in the aqueous phase. A plot of the % protonation (Fig. 2) shows that the limited solubility of HA dramatically increases the pH of the midpoint. Thus the pH of the midpoint is greater than the pKa for an anionic surfactant if [A−]0 > 2S0. For dodecanoic acid, the pH of the midpoint is 8.3, 7.3, or 6.6 for initial concentrations ([A−]0) of 50 mM, 5 mM, and 1 mM, respectively. Our results (Table 1) show that for a 5 mM solution of sodium dodecanoate, the pH of the solutions in the presence and absence of CO2 are ∼5.8 and 8.3, which is consistent with the predicted midpoint of approximately 7.3. Hence it is not necessary for the pH to drop below the pKa in order to convert an anionic surfactant to the neutral form; a drop below the solubility-modified midpoint is sufficient.
 | (6) |
2.3 Washing oil-contaminated sand with switchable and nonswitchable surfactants
We compared three SAS (compounds 1a, 3a, and 4a) with two non-switchable surfactants (Scheme 2): an alkylsulfate (sodium dodecylsulfate (SDS)) and a neutral surfactant (Triton X-100). SDS is a fairly ubiquitous anionic surfactant used in a variety of products, and has been reported as a common reference surfactant.35 Triton X-100 is a non-ionic, biodegradable surfactant that is one of the family of alkylphenol “polyethoxylates” (RC6H4(OC2H4)xOH) which have been reported in the soil washing literature.2,36 The benzoate surfactant 3a was chosen to determine whether an aromatic ring would make the surfactant more effective compared to aliphatic carboxylates in terms of solubilization of oil. Because the purpose of our initial experiments was neither to optimize the process nor to determine the best surfactant, but rather to demonstrate whether switchable surfactants are capable of soil washing, we tested only a single concentration of surfactant; 1 wt% was chosen because it is above the CMC for all the surfactants studied (Table 2). | ||
| Scheme 2 The two surfactants used in this study that are non-switchable (not CO2-responsive). | ||
| Surfactant | MW (g mol−1) | Solubility (g L−1) | CMC (mM) | CMC (g L−1) | CMC wt% |
|---|---|---|---|---|---|
| a Determined experimentally. See ESI. | |||||
| SDS | 288.4 | 150 (ref. 37) | 8.0 (ref. 38) | 2.3 | 0.23 |
| Triton X-100 | 625 | Soluble39,40 | 0.24 (ref. 39) | 0.15 | 0.015 |
| 1a | 318.1 | N/A | 2.8a | 0.89 | 0.089 |
| 3a | 272.3 | N/A | 14a | 3.8 | 0.38 |
| 4a | 222.3 | 12 (ref. 41) | 24 (ref. 42) | 5.3 | 0.53 |
Samples of oil-contaminated soil were prepared artificially. To simplify this initial study, Ottawa Sand was chosen as a model “soil” matrix for the washing studies. Ottawa Sand is a nonporous silica sand which contains little organic matter and has a particle size range of typically 0.1 to 1.0 mm.43,44 Our sample had a particle size range of 0.5 to 1.5 mm, measured by optical spectroscopy. Future work will need to evaluate the effect of different sands and soils on the process performance. The sand was artificially contaminated with a light crude, North Sea crude oil (properties given in Table S1†).
The contaminated sand was weathered for these studies. Weathering is an important process in the artificial contamination of the sand because it better mimics real environmental samples. In real soil washing applications, the soil has been weathered by exposure to ambient conditions for an extended time, which increases the binding of the contaminants to the soil matrix and makes remediation more difficult.35
Various batches of contaminated and artificially-weathered sand were created for this work (Table S2†), containing 2.6 to 3.1 wt% oil after the weathering. The average percent mass of oil lost during weathering was 26%.
Soil washing experiments were done at room temperature (23 °C) and at elevated temperature (50 °C). The room temperature experiments were intended to determine the surfactants' performance without the aid of heating, which should maximize the difference in washing performance between surfactants. Furthermore if the switchable surfactants performed well at room temperature, it would increase the desirability of these chemicals because their usage could potentially reduce the energy cost of soil washing. Elevated temperatures increased the efficiency of soil washing, as expected, because solubility and desorption kinetics are increased, and the viscosity of the oil is reduced which helps to solubilize the oil.4,35
Contaminated and weathered sand samples (5 g) were washed with 10 g solutions of surfactant (Fig. 3). Typically, two or three distinct layers are apparent after washing: (1) sand on the bottom, (2) an opaque, tan coloured aqueous layer, and (3) in some cases, a thin oil layer was observed on the top. All surfactants removed at least some oil from the sand during the washing. In contrast, water without surfactant was unable to remove a significant amount of oil. Of the five surfactants, 1a visually appeared to remove the most oil from the sand (Fig. S5†).
 | ||
| Fig. 3 The sand washing process at room temperature. | ||
After the washing was completed, the liquid phase of the wash mixture was decanted into a separate vial (first decant). The sand was then rinsed with 50 mL of deionized water. The oil contents of the washed sand, the decanted surfactant solution, and the rinse solution were all determined.
The amount of oil remaining on the sand after washing was determined by organic solvent extraction of the residual oil using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v:v mixture of dichloromethane and hexanes. At room temperature, the switchable surfactant 1a and the non-switchable surfactant Triton X-100 were reasonably effective (Fig. 4). For all surfactants, the oil removal from the sand was visibly more complete and faster at 50 °C than at room temperature, as expected. At 50 °C, all of the switchable and non-switchable surfactants were reasonably effective.
:1 v:v mixture of dichloromethane and hexanes. At room temperature, the switchable surfactant 1a and the non-switchable surfactant Triton X-100 were reasonably effective (Fig. 4). For all surfactants, the oil removal from the sand was visibly more complete and faster at 50 °C than at room temperature, as expected. At 50 °C, all of the switchable and non-switchable surfactants were reasonably effective.
 | ||
| Fig. 4 Comparison of the average% removal of oil from sand by the surfactants after sand washings at room temperature (white) and 50 °C (grey). Error bars represent the standard deviation (n = 3). | ||
2.4 Processing of the decanted wash mixture
The purpose of using a switchable surfactant is to facilitate the recovery of the contaminant and the surfactant. In typical soil washing operations using conventional surfactants, the wash mixture (emulsified oil in water) is generally sent for further treatment, including by aeration, filtration, adsorption using activated carbon, reverse osmosis and electrodialysis.45,46 These treatments will recover the water, but the surfactant and contaminant are non-recoverable due to biodegradation or subsequent treatments, along with the difficulty of separating them. Both the contaminant and the surfactant are potentially valuable components and, if recovered and re-used, waste could be reduced and revenue generated to offset the cost of remediation. When a switchable surfactant is turned off it may destabilize (break) the emulsion.Emulsions stabilized by cationic or anionic surfactants can be broken by increasing the ionic strength by addition of a salt. While this process is effective and fairly simple, there are drawbacks, including the cost of purchasing the salt and treating and disposing of the salty aqueous waste. We found that the SDS wash solution emulsions could be broken by adding NaCl until a distinct oil/water phase separation was observed. The samples were left to settle overnight, but separation began almost immediately after salt addition. Regardless of whether the washing had been performed at room temperature or 50 °C, separation by salt addition was achieved; there was no oil visually evident in the clear and colourless water layer, and the mixture could not be re-emulsified by shaking or stirring. However, the salt addition also caused the SDS to precipitate, and the oil adhered to the solid surfactant particles to make a tan-coloured slurry layer above the water. Salt addition did not cause the separation of the wash solutions containing Triton X-100.
Even without addition of salt, the wash solution emulsions partially settled out. Some of the oil creamed to form a thin layer above the bulk emulsion. If left overnight, the amount of oil in the aqueous phase dropped by 70% for both SDS and Triton X-100. When CO2 was bubbled through the emulsion immediately after washing, the amount of creaming was the same, and the amount of oil remaining in the aqueous phase also dropped by 70%. This demonstrated that CO2 does not help break the wash solution emulsions when non-switchable surfactants are used. In all cases, this separation by creaming is not permanent; even a mild disturbance of the vials caused the creamed oil to re-enter the emulsion.
The wash solution emulsions containing switchable surfactants were effectively broken by the addition of CO2. The reduction in pH caused the precipitation of the surfactant in its neutral form, causing the mixture to become visibly turbid. After 10 min of bubbling CO2, precipitation of the surfactant was complete; foaming ceased and the components (oil, water and surfactant) no longer formed an emulsion. Separation of the phases did not occur, however, as rapidly as it did in samples with SDS treated with NaCl. The treated wash mixtures were left to stand overnight, during which time the separation of the crude oil and surfactant from water occurred (Tables 3 and 4). While CO2 treatment was always done at room temperature, the quality of the separation was better if the washing step had been performed at 50 °C than at room temperature. Particularly with 1a and 4a, the oil contamination remaining in the aqueous phase was nearly as low after the CO2 and settling treatment as it was with SDS after NaCl treatment. The switchable surfactants allowed the oil to be separated much more effectively than neutral Triton X-100, though slightly less effectively than SDS. The switchable surfactants used the benign and easily removed trigger CO2, so that the water would not require expensive desalination before reuse.
| Surfactant | Treatment | goil/Laq before treatment | goil/Laq after treatment | % Removal of oil from aq. phase |
|---|---|---|---|---|
| a % Removal = {1 − (oil mass after CO2)/(oil mass before CO2)} × 100%. | ||||
| SDS | NaCl | 1.7 | 0.35 | 79 |
| X-100 | NaCl or CO2 | 5.8 | — | None |
| 1a | CO2 | 2.6 | 0.48 | 81 |
| 3a | CO2 | 2.2 | 1.0 | 55 |
| 4a | CO2 | 1.4 | 0.63 | 55 |
| Surfactant | Treatment | goil/Laq before treatment | goil/Laq after treatment | % Removal of oil from aq. phase |
|---|---|---|---|---|
| a % Removal = {1 − (oil mass after CO2)/(oil mass before CO2)} × 100%. | ||||
| SDS | NaCl | 9.8 | 0.25 | 98 |
| X-100 | NaCl or CO2 | 11 | — | None |
| 1a | CO2 | 4.4 | 0.32 | 93 |
| 3a | CO2 | 6.7 | 0.73 | 89 |
| 4a | CO2 | 11 | 0.5 | 95 |
2.5 Fate of the oil and surfactant
The overall performance of the soil washing for the optimal case, using surfactant 1a at 50 °C, is summarized by tracking the average amount of the oil in the various compartments as shown in Fig. 5. The initial wash mixture contained 78% of the oil originally on the sand after careful decanting. Another 13% of the oil was removed with the water rinse step, indicating that closer to 90% could likely be removed in the initial wash with a more efficient separation method. The final “cleaned” sand had 3% of the initial oil remaining (i.e. the contamination of the sand by oil decreased from 30000 mg kg−1 before washing to 800 mg kg−1 afterwards). The initial wash mixture was kept separate from the rinse to maximize the efficiency of further recovery. Of the 78% of the oil in the wash mixture, 73% was isolated in an oil layer after the surfactant was switched off, with the other 5% remaining in the wash water. The potential for recovering a significant portion of the oil removed from the sand has been demonstrated, though the recovered oil may need further treatment before it can be used.
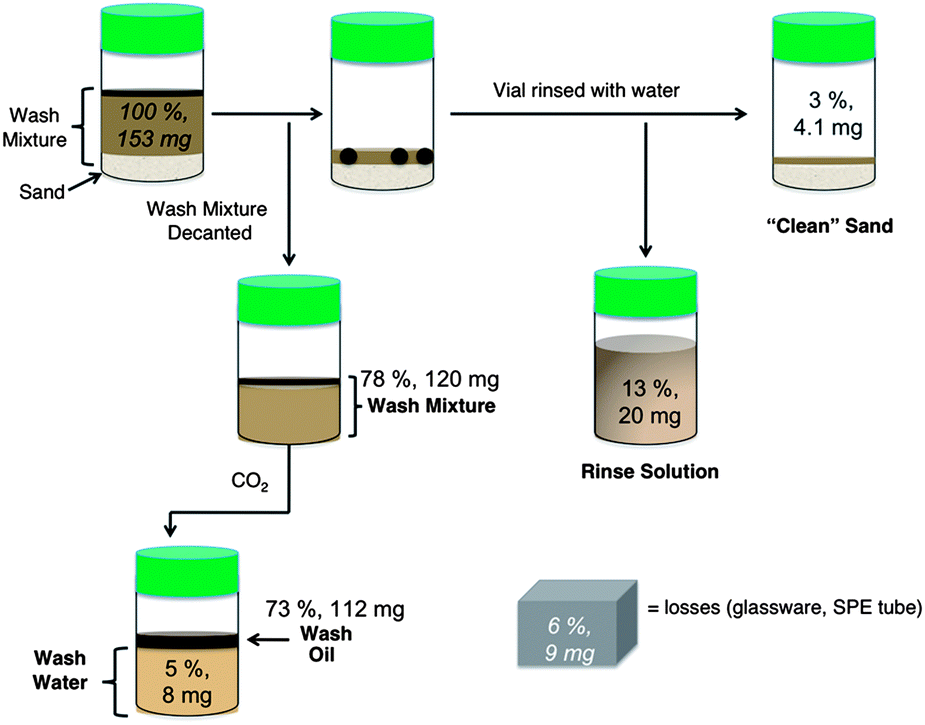 | ||
| Fig. 5 Average percent oil in various compartments after the washing of contaminated sand using surfactant 1a at 50 °C. | ||
Further work is needed to determine the fate of the surfactant throughout the wash process. The residual surfactant on the cleaned sand was about 0.45%, while the amount in the wash water, after precipitation of the 1b form, was 0.48%. We were not able to separate effectively the precipitated 1b from 1b dissolved in the oil layer due to the small scale of our experiments. A larger scale operation with specialized handling equipment would be able to recover and then reuse some of the surfactant, though recovery from the wash oil layer will require development of additional separation procedures.
Conclusions
Carboxylate and phenolate anionic surfactants can be deactivated and precipitated by the addition of an atmosphere of CO2, which lowers the pH below the midpoint of the surfactant (the pH corresponding to 50% protonation). The pH required to destabilize emulsions is not as low as the pKa; the limited solubility of the neutral form of the surfactant causes the midpoint to move to significantly higher pH values.The switchable surfactants presented are able to remove crude oil from contaminated sand and afford subsequent separation of the crude oil from the water upon CO2 treatment. At room temperature, switchable surfactant 1a and non-switchable surfactant Triton X-100 had reasonable oil-removable performance, while at 50 °C all of the switchable surfactants and Triton X-100 removed at least 90% of the oil from contaminated sand, while non-switchable surfactant SDS performed nearly as well.
After washings at 50 °C with switchable surfactants, treatment of the wash solutions with CO2 gas triggered the precipitation of the surfactant and the breaking of the emulsion, lowering the concentration of oil in the water by up to 95% and lowering the concentration of surfactant in the water by up to 99.5% relative to the original concentration.
In contrast, washing the sand with aqueous solutions of the non-switchable surfactant SDS removed less oil from the sand and gave emulsions that could not be broken by CO2 addition, although salt addition did break the emulsions. Washing the sand with aqueous solutions of the non-switchable surfactant Triton X-100 gave emulsions that could not be broken by either CO2 or salt addition.
Experimental methods
Experimental methods are described in the ESI.† The procedure for the washing of oil-contaminated sand is also shown here for the convenience of the reader.All sand washings were done in triplicate. Approximately 5.0 g of the contaminated sand, 10 g of the desired surfactant solution, and a stir bar were placed into a 20 mL vial, which was then capped with a poly(tetrafluoroethylene)-lined lid. The samples were shaken by hand for 20 s, placed in an otherwise empty 150 mL beaker (for support) and then stirred at 470 rpm for 1 h, re-shaken for another 20 s and stirred for another hour. For the washings at 50 °C, the samples were placed directly into the water bath, which was previously heated to 50 °C. After the 2 h, the samples were left to stand briefly to let the solution settle. Fig. S4† illustrates the sand washing process and Fig. S5† shows photographs of some of the samples at the start and end of the 2 h washing period.
After the washing was completed, the liquid phase of the wash mixture was decanted into a separate 20 mL vial and the mass was recorded. This initial decanted solution will be referred to as the first decant. The sand was then rinsed with 50 mL of deionized water and the rinses were combined in a 100 mL Wheaton jar and are referred to as the rinse solution. The rinse solution was analyzed for oil content as described in section 1.7.3 of the ESI.†
A small sample of the decant solution was removed for analysis (see section 1.7.2 of the ESI†). The vial containing the remainder of the decant was capped with a rubber septum. CO2 was bubbled into the vial using a syringe needle at a flow rate of 3 to 5 mL min−1 for 10 min. A separate syringe needle was used as a vent. After 10 min, the vial was re-capped with a lid and left to settle overnight, after which the aqueous phase was again analyzed for oil content.
Acknowledgements
The authors gratefully thank the Natural Sciences and Engineering Research Council of Canada and Albonia Innovative Technologies for funding. PGJ also acknowledges support from the Canada Research Chairs Program.Notes and references
- L. K. Wang, in Site remediation and groundwater decontamination, CRC Press, 2007 Search PubMed
.
- S. Deshpande, B. J. Shiau, D. Wade, D. A. Sabatini and J. H. Harwell, Water Res., 1999, 33, 351–360 CrossRef CAS
- M. J. Mann, J. Hazard. Mater., 1999, 66, 119–136 CrossRef CAS
- M. I. Kuhlman and T. M. Greenfield, J. Hazard. Mater., 1999, 66, 31–45 CrossRef CAS
- H. Sakai, A. Matsumura, S. Yokoyama, T. Saji and M. Abe, J. Phys. Chem. B, 1999, 103, 10737–10740 CrossRef CAS
- A. F. Dexter, A. S. Malcolm and A. P. J. Middelberg, Nat. Mater., 2006, 5, 502–506 CrossRef CAS PubMed
- C. B. Minkenberg, L. Florusse, R. Eelkema, G. J. M. Koper and J. H. v. Esch, J. Am. Chem. Soc., 2009, 131, 11274–11275 CrossRef CAS PubMed
- T. Saji, K. Hoshino and S. Aoyagui, J. Am. Chem. Soc., 1985, 107, 6865–6868 CrossRef CAS
- P. Anton, P. Koeberle and A. Laschewsky, Prog. Colloid Polym. Sci., 1992, 89, 56–59 CAS
- N. Aydogan and N. L. Abbott, Langmuir, 2001, 17, 5703–5706 CrossRef CAS
- S. S. Datwani, V. N. Truskett, C. A. Rosslee, N. L. Abbott and K. J. Stebe, Langmuir, 2003, 19, 8292–8301 CrossRef CAS
- K. Tsuchiya, Y. Orihara, Y. Kondo, N. Yoshino, T. Ohkubo, H. Sakai and M. Abe, J. Am. Chem. Soc., 2004, 126, 12282–12283 CrossRef CAS PubMed
- H. Sakai and M. Abe, in Mixed Surfactant Systems, ed. M. Abe and J. F. Scamehorn, Marcel Dekker, 2nd edn, 2005, New York, pp. 507–543 Search PubMed
- M. M. Schmittel, M. Lal, K. Graf, G. Jeschke, I. Suske and J. Salbeck, Chem. Commun., 2005, 5650–5652 RSC
- Z. Cheng, B. Ren, M. Gao, X. Liu and Z. Tong, Macromolecules, 2007, 40, 7638–7643 CrossRef CAS
- S. Ghosh, K. Irvin and S. Thayumanavan, Langmuir, 2007, 23, 7916–7919 CrossRef CAS PubMed
- Y. Liu, P. G. Jessop, M. Cunningham, C. A. Eckert and C. L. Liotta, Science, 2006, 313, 958–960 CrossRef CAS PubMed
- L. Scott, T. Robert, J. R. Harjani and P. G. Jessop, RSC Adv., 2012, 2, 4925–4931 RSC
- C. I. Fowler, C. Muchemu, R. E. Miller, L. Phan, C. O'Neill, P. G. Jessop and M. F. Cunningham, Macromolecules, 2011, 44, 2501–2509 CrossRef CAS
- C. I. Fowler, P. G. Jessop and M. F. Cunningham, Macromolecules, 2012, 45, 2955–2962 CrossRef CAS
- C. Liang, J. R. Harjani, T. Robert, E. Rogel, D. Kuehne, C. Ovalles, V. Sampath and P. G. Jessop, Energy Fuels, 2012, 26, 488–494 CrossRef CAS
- W. Chu, Pract. Period. Hazard., Toxic, Radioact. Waste Manage., 2003, 7, 19–24 CrossRef CAS
- A. Lottermoser, Trans. Faraday Soc., 1934, 31, 200–204 RSC
- E. R. Moore and N. A. Lefevre, US Pat., 4,623,678, 1986
- E. Penth, US Pat., 5,435,920, 1995
- V. Nyren and E. Back, Acta Chem. Scand., 1958, 12, 1305–1311 CrossRef CAS PubMed
- M. Rapoport, C. K. Hancock and E. A. Meyers, J. Am. Chem. Soc., 1961, 83, 3489–3494 CrossRef CAS
- Determination of Organic Structures by Physical Methods, ed. E. A. Braude and F. C. Nachod, Academic Press, 1955 Search PubMed
- R. A. Peters, Proc. R. Soc. London, Ser. A, 1931, 133, 140–154 CrossRef CAS
- R. W. Wakelin, Trans. Faraday Soc., 1938, 34, 1535–1536 RSC
- K. A. Coltharp and E. I. Franses, Colloids Surf., A, 1996, 108, 225–242 CrossRef CAS
- S. S. Tzocheva, P. A. Kralchevsky, K. D. Danov, G. S. Georgieva, A. J. Post and K. P. Ananthapadmanabhan, J. Colloid Interface Sci., 2012, 369, 274–286 CrossRef CAS PubMed
- N. T. Hansen, I. Kouskoumvekaki, F. S. Jørgensen, S. Brunak and S. Ó. Jónsdóttir, J. Chem. Inf. Model., 2006, 46, 2601–2609 CrossRef CAS PubMed
- A. Llinàs, J. C. Burley, K. J. Box, R. C. Glen and J. M. Goodman, J. Med. Chem., 2007, 50, 979–983 CrossRef PubMed
- K. Urum, T. Pekdemir and M. Çopur, J. Colloid Interface Sci., 2004, 276, 456–464 CrossRef CAS PubMed
- M. J. Rosen, Surfactants and Interfacial Phenomena, Wiley, Hoboken, NJ, 3rd edn, 2004 Search PubMed
- K. Verschueren, Handbook of environmental data on organic chemicals, Wiley, New York, 4th edn, 2001 Search PubMed
- A. Dominguez, A. Fernández, N. González, E. Iglesias and L. Montenegro, J. Chem. Educ., 1997, 74, 1227–1231 CrossRef CAS
- Triton X-100 Product Information, Sigma Aldrich Chemical Company, 1999 Search PubMed.
- TritonTM X-100 Surfactant, Form No. 119-01882-1207, The Dow Chemical Company, 2012 Search PubMed.
- L. Shedlovsky, G. D. Miles and G. V. Scott, J. Phys. Colloid Chem., 1947, 51, 391–407 CrossRef CAS
- A. N. Campbell and G. R. Lakshminarayanan, Can J. Chem., 1965, 43, 1729–1737 CrossRef CAS
- R. Salgado, P. Bandini and A. Karim, J. Geotech. Geoenviron. Eng., 2000, 126, 451–462 CrossRef
- K. D. Pennell, L. M. Abrlola and W. J. Weber Jr, Environ. Sci. Technol., 1993, 27, 2332–2340 CrossRef CAS
- C. Baird, Environmental Chemistry, W. H. Freeman, New York, 2nd edn, 1998 Search PubMed
- Environmental Engineering, ed. N. L. Nemerow, F. J. Agardy, P. Sullivan and J. A. Salvato, John Wiley & Sons, Hoboken, New Jersey, 2009 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, synthesis and characterization of compounds 1b and 2b, characterization and properties of the North Sea crude oil, emulsion stability tests, oil content of sand before and after weathering. See DOI: 10.1039/c3ra47158f |
| This journal is © The Royal Society of Chemistry 2014 |