DOI:
10.1039/C3RA47129B
(Paper)
RSC Adv., 2014,
4, 6417-6423
Asymmetric transfer hydrogenation of unsymmetrical benzils†
Received
28th November 2013
, Accepted 18th December 2013
First published on 20th December 2013
Abstract
In this paper, the asymmetric transfer hydrogenation of unsymmetrical benzils with m, p-substituents was conducted with a substrate/catalyst molar ratio of 100 at 40 °C for 24 h to produce (S,S)-hydrobenzoins in good yields (76.2% to 97.1%) with high diastereomeric (syn/anti = 10.8 to 29.7/1) and enantiomeric purities (86.1%ee syn to 98.9%ee syn). Unfortunately, the unsymmetrical benzils with the o-substituents such as electron-donating (R = CH3, OCH3) and electron-withdrawing groups (R = F, Cl, CF3) resulted in poor yields (0% to 31.2%), even at 40 °C for 72 h. These products had inefficient diastereoselectivities (syn/anti = 1.5 to 5.0/1) caused by steric effects. Furthermore, the results of a dynamic–kinetic study were used to propose a plausible reaction pathway of unsymmetrical benzil using 3-methoxy-1,2-diphenyl ethanedione as an example.
Introduction
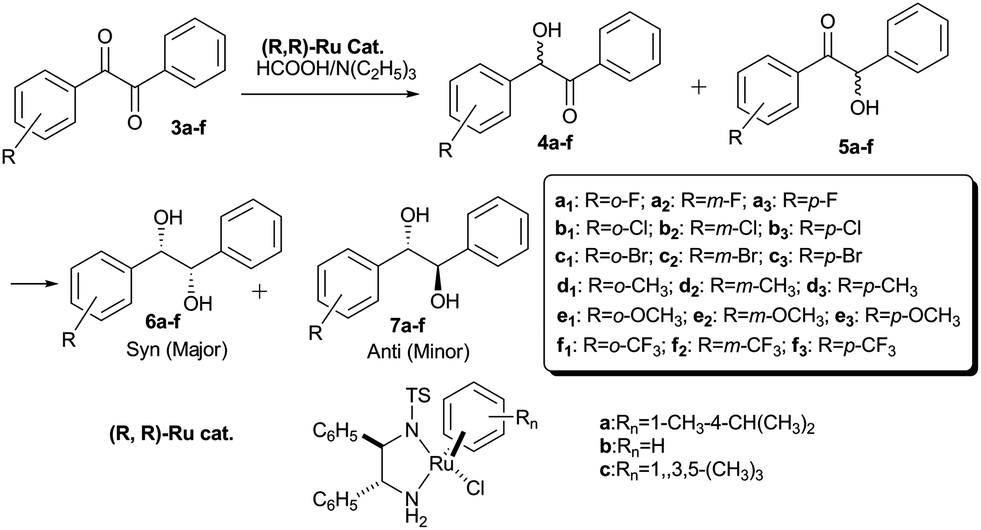
Chiral 1,2-diols (especially, hydrobenzoins) are useful building materials for the syntheses of various biologically active compounds, as well as chiral ligands and auxiliaries in stereoselective organic syntheses.1 These important chiral hydrobenzoins have been prepared through the resolution of racemic hydrobenzoins,2 stereoselective pinacol coupling,3 Sharpless asymmetric dihydroxylation,4 and asymmetric transfer hydrogenation.5 As early as 1999, T. Ikariya et al. developed a practical stereoselective synthesis of chiral hydrobenzoins via asymmetric transfer hydrogenation of symmetrical benzils.5d Until now, there was no report available on the asymmetric transfer hydrogenation of unsymmetrical benzils to optically active hydrobenzoins, with regard to unavailable raw materials, separation of four stereoisomers and absolute configuration assignment. In the current paper, we developed asymmetric synthesis of optically active hydrobenzoins via the asymmetric transfer hydrogenation of unsymmetrical benzils catalyzed by the well-defined RuCl(R,R)-Tsdpen(η6-arene) chiral catalysts (a–c) (Scheme 1).6 Furthermore, the dynamic–kinetic study on the reactant, intermediates and products, which was monitored through chiral high-performance liquid chromatography (HPLC), provided direct insight into asymmetric hydrogenation mechanism of unsymmetrical benzils to 1,2-diols.
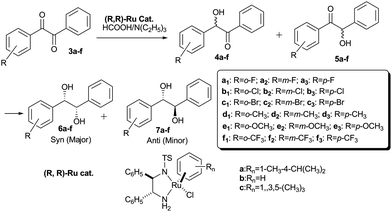 |
| | Scheme 1 Asymmetric transfer hydrogenation of unsymmetrical benzils. | |
Results and discussion
Synthesis of unsymmetrical benzils
1,3-Diketones 2a–f in the enol form that were verified through 1H NMR were prepared by the reaction of substituted acetophenone with ethyl benzoate from moderate to high yields (40% to 92%). The selective cleavage of the C–C bonds of 1,3-diketones 2a–f produced the useful unsymmetrical benzils 3a–f in 35% to 90% yields at 30 °C for 12 h using a combination of 20 mol% of FeCl3 and 500 mol% tert-butyl nitrite.7 Fortunately, various 1,3-diketones 2a–f, with both the electron-donating (R = CH3 and OCH3) and electron-withdrawing (R = F, Cl, Br and CF3) substituents in the o-, m- and p-position of aryl ring, tolerated the reaction conditions well to produce the corresponding unsymmetrical benzils 3a–f (Scheme 2).
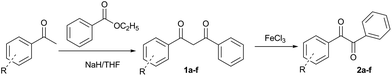 |
| | Scheme 2 The synthetic route to various unsymmetrical benzils 3a–f. | |
Optimization of catalytic conditions
The catalytic conditions in the asymmetric transfer hydrogenation of unsymmetrical benzils 3a–f catalyzed by RuCl (R,R)-Tsdpen(η6-p-cymene) were screened out, and some representative examples are listed in Table 1. Chiral (R,R)-Ru(II) a with arene ligand η6-p-cymene [Rn = 1-CH3 and 4-CH(CH3)2] showed the best catalytic performances (98.8%ee syn, Dr (syn/anti) = 28.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and 94.9% yield, entry 1, Table 1); whereas the other benzene complexes b (Rn = H), c [Rn = 1,3,5-(CH3)3] were less reactive. The molar ratio of HCO2H to NEt3 had an important function in catalytic performance. The best catalytic results of 3e2 in terms of yield and stereoselectivity were obtained at the 4:2 molar ratio of HCO2H to NEt3. Surprisingly, the 1,2-diol 6e2 yields were apparently decreased to low to moderate yields (0–74.6%) in various organic solvents. However, some improved %ee syn (>99%) and Dr (syn/anti > 30/1) values were found in acetone, DMF, and acetonitrile (entries 4–9, Table 1). Therefore, it was concluded that organic solvent was not crucial for chiral (R,R)-Ru(II) catalyst a to be efficient and practical. At relatively higher temperatures (e.g., 60 °C and 80 °C), the enantioselectivity (98.8%ee syn → 97.2%ee syn) and diastereoselectivity (38.9/1 → 10.4/1) decreased slightly (entries 10–14, Table 1). The molar ratio of substrate to catalyst (S/C) critically affected the 1,2-diol yields. Unsymmetrical benzil 3e2 with a S/C below 200 can produce more than 90% yields with an excellent enantioselectivity (98.6%ee syn to 99.4%ee syn) and diastereoselectivity (syn/anti = 21.8/1 to 28.3/1) (entries 15–18, Table 1). Fortunately, the excellent stereoselectivity (98.4%ee syn) and diastereoselectivity (syn/anti = 39.7) with the less reactivity (58.3%) can be obtained, even with a S/C = 500 at 40 °C for 24 h (Table 1, entry 19).
:1, and 94.9% yield, entry 1, Table 1); whereas the other benzene complexes b (Rn = H), c [Rn = 1,3,5-(CH3)3] were less reactive. The molar ratio of HCO2H to NEt3 had an important function in catalytic performance. The best catalytic results of 3e2 in terms of yield and stereoselectivity were obtained at the 4:2 molar ratio of HCO2H to NEt3. Surprisingly, the 1,2-diol 6e2 yields were apparently decreased to low to moderate yields (0–74.6%) in various organic solvents. However, some improved %ee syn (>99%) and Dr (syn/anti > 30/1) values were found in acetone, DMF, and acetonitrile (entries 4–9, Table 1). Therefore, it was concluded that organic solvent was not crucial for chiral (R,R)-Ru(II) catalyst a to be efficient and practical. At relatively higher temperatures (e.g., 60 °C and 80 °C), the enantioselectivity (98.8%ee syn → 97.2%ee syn) and diastereoselectivity (38.9/1 → 10.4/1) decreased slightly (entries 10–14, Table 1). The molar ratio of substrate to catalyst (S/C) critically affected the 1,2-diol yields. Unsymmetrical benzil 3e2 with a S/C below 200 can produce more than 90% yields with an excellent enantioselectivity (98.6%ee syn to 99.4%ee syn) and diastereoselectivity (syn/anti = 21.8/1 to 28.3/1) (entries 15–18, Table 1). Fortunately, the excellent stereoselectivity (98.4%ee syn) and diastereoselectivity (syn/anti = 39.7) with the less reactivity (58.3%) can be obtained, even with a S/C = 500 at 40 °C for 24 h (Table 1, entry 19).
Table 1 Asymmetric hydrogenation of benzil 3e2 under various conditionsa
Asymmetric transfer hydrogenation of various unsymmetrical benzils
The optimized protocol was expanded to a wide variety of various unsymmetrical benzils 3a–f with o-, m- and p-substituents on aromatic rings to investigate catalytic performance of (R,R)-Ru(II) catalyst a. Based on the results in Table 2, the transfer hydrogenation of unsymmetrical benzils was closely related to the presence of o-, m- and p-substituting position on phenyl group. When the substituents, including electron-donating (R = CH3 and OCH3) and electron-withdrawing groups (R = F and Cl) groups, were in the ortho-position on phenyl group, unsymmetrical benzils 3a1–e1 were reduced with the low reactivities (0% to 31.2% yields) and reduced diastereoselectivities (syn/anti = 1.3 to 5.0) at 40 °C for 72 h; these results were caused by sterically hindered interaction between the o-position substituent and carbonyl group. However, the excellent enantioselectivities (92.0%ee syn to 97.4%ee syn) of starting materials 3a1–e1 were observed (entries 1, 4, 10 and 13, Table 2). Our attempted reduction of unsymmetrical benzil 3f1 with a bulky and strong electron-withdrawing o-CF3 group under the same conditions only produced the intermediates 4f1 and 5f1 with low % ees and yields. Moreover, hydrobenzoin 6f1 was not obtained. Especially, benzil 3c1 with the o-Br substituent gave the excellent stereoselectivity (98.7%ee syn, syn/anti = 21.6) owing to the suitable volume of bromine atom. Therefore, it could be concluded that the bulkier o-position substituent resulted in the slower reactivity, even hydrogenation reaction could not proceeded smoothly.
Table 2 Asymmetric transfer hydrogenation of various unsymmetrical benzilsa
Fortunately, the various unsymmetrical benzils 3a2,3–f2,3 with the m-, p-substituents attached to the phenyl group, both electron-donating (R = CH3 and OCH3) and electron-withdrawing (R = F, Cl, Br and CF3) substituents, can be stereoselectively reduced to optically active (S,S)-hydrobenzoins 6a2,3–f2,3. These products exhibited good to excellent enantioselectivities (86.1%ee syn to 98.9%ee syn) and diastereoselectivities (syn/anti = 10.8 to 29.7) in 76.2% to 97.1% yields in HCOOH/N(C2H5)3 = 2:1 with a S/C molar ratio of 100 at 40 °C for 24 h. Notably, a pair of (S,S) and (R,R)-hydrobenzoins 6b2, 6c2, 6e3 enantiomers were difficult to be isolated through chiral HPLC equipped with current commercial chromatographic columns (entries 5, 8, and 15, Table 2).
Dynamic kinetic study
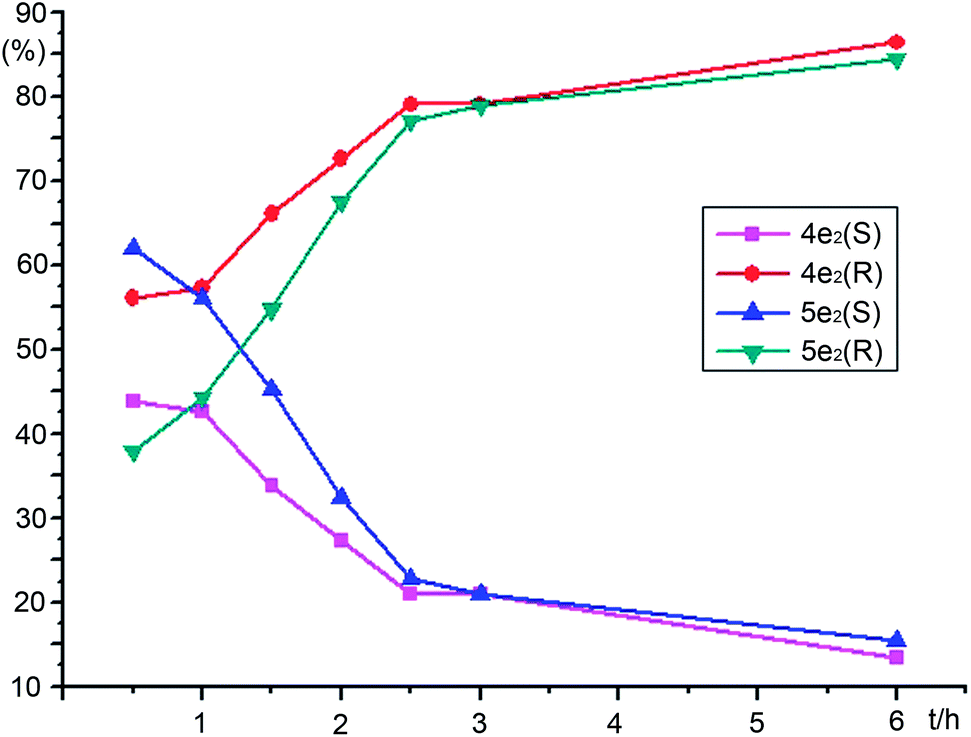
First, to identify the intermediates, two racemic α-hydroxy ketones 4e2 and 5e2 were synthesized according to the reference materials (Scheme 3).8 To elucidate the hydrogenation mechanism of unsymmetrical benzil, e.g., 3e2 with m-OCH3 substituent, the stereoselectivities and reactivities during the course of catalytic reaction were monitored using HPLC. Based on the peak areas in chiral HPLC, the reactivity and enantioselectivity profiles of benzil 3e2, intermediates 4e2 and 5e2, and hydrobenzoin 6e2 were plotted versus time during the whole process; the plots are shown in Fig. 1 and 2, respectively.
 |
| | Scheme 3 Synthesis of racemic α-hydroxy ketones 4e2 and 5e2. | |
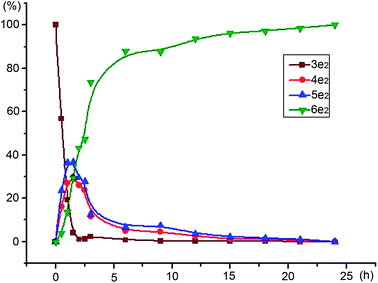 |
| | Fig. 1 The reactivity profiles of benzil, intermediates and 1,2-diol plotted versus time during the experiment. | |
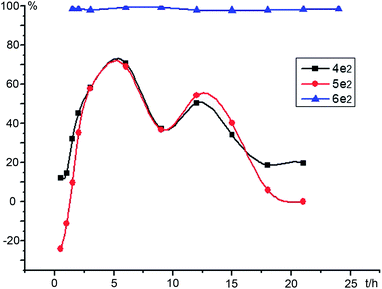 |
| | Fig. 2 The enantioselectivity profiles of benzil, intermediates and 1,2-diol plotted versus time during the experiment. | |
From Fig. 1, the conversion of benzil 3e2 to α-hydroxy ketones 4e2 and 5e2 was completed in 2 h at 40 °C; and most of α-hydroxy ketones (>90%) were converted to (S,S)-hydrobenzoin 6e2 within 12 h. A few points were noted that need further investigation. First, the regioselective reduction of unsymmetrical benzil 3e2 to α-hydroxy ketones 4e2 and 5e2 was not observed as expected. In addition, the intermediate 4e2 was less abundant than 5e2, although two carbonyl functional groups possessed the different cloud densities caused by the electron effect of electron-donating m-OCH3 group. Second, the excellent enantioselectivities of (S,S)-6e2 (>98%ee) had been maintained at a high level from beginning to end during the entire catalytic process and were not influenced by the enantioselectivities of 4e2 and 5e2; although, the low to moderate %ee values of 4e2 (10%ee to 70%ee) and 5e2 (−25%ee to 70%ee) changed in the action of oscillation process (Fig. 2). In particular, the enantioselectivity of intermediate 5e2 changed from negative to positive values at about 2 h. Therefore, (R)-enantiomers 4e2 and 5e2 gradually predominated over their (S)-enantiomers, and proceeded more slowly than their enantiomers (S)-4e2 and 5e2 (Fig. 3). Theoretically, (R)-4e2 and 5e2 can be hydrogenated sequentially to two possible stereoisomers, (R,R)-6e2 and (R,S)-7e2. Unfortunately, small amounts of (R,R)-6e2 and (R,S)-7e2 were observed in the resulting products monitored through HPLC. In fact, whether 4e2 and 5e2 were (R) or (S)-configuration, these enantiomers produced (S,S)-hydrobenzoin 6e2 with an excellent stereoselectivity of 98.9%ee syn and syn/anti = 29.7. The events that occurred with regard to intermediates (R)-4e2 and 5e2 need to be discussed.
 |
| | Fig. 3 The relative contents of (S)-4e2, 5e2 and (R)-4e2, 5e2 in asymmetric hydrogenation of benzil 3e2 over 6 h. | |
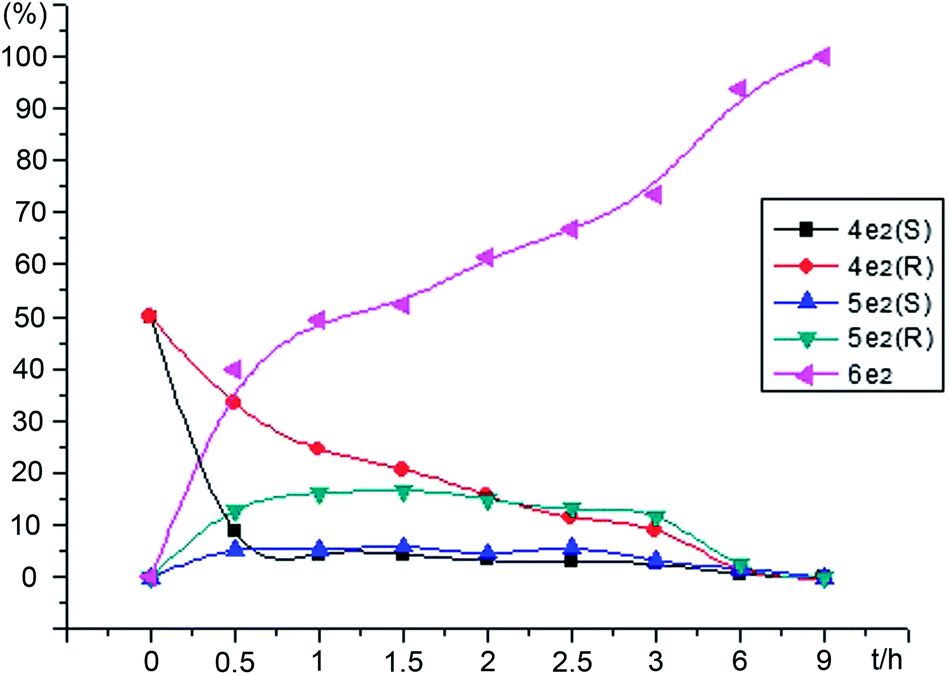
To investigate how (R) and (S)-4e2 and 5e2 were converted to 1,2-diols, the racemic α-hydroxy ketones 4e2 and 5e2 were hydrogenated independently with a S/C molar ratio of 100 in HCOOH/N(C2H5)3 = 2:1 at 40 °C, and were monitored via HPLC. As shown in Fig.4, in the hydrogenation of racemic 4e2, the conversion of (S)-4e2 was three to five times faster than that of (R)-4e2 within 3 h. In particular, the (R) and (S)-5e2 intermediates were unexpectedly found, and the (S)-5e2 content was also lower than (R)-5e2. Likewise, (R) and (S)-4e2 intermediates were also observed in the hydrogenation of racemic 5e2 (also see ESI†). Therefore, based on these findings and final (S,S)-6e2 product, the slow-reacting (R)-isomers were subjected to rapid racemization among (R)-5e2, (S)-5e2, (S)-4e2, and (R)-4e2 intermediates through keto–enol tautomerism equilibrium; and the intermediates (S)-5e2 and (S)-4e2 were hydrogenated asymmetrically to (S,S)-6e2 in 94% yield with syn/anti = 29.7/1 and 98.9%ee syn. Otherwise, (R,R)-6e2 and (R,S)-7e2 can be formed by the direct hydrogenation of intermediates (R)-4e2 and (R)-5e2. Therefore, a plausible reaction pathway for the catalytic asymmetric transfer hydrogenation of unsymmetrical benzil 3e2 was proposed in Scheme 4.
 |
| | Fig. 4 The reactivity profiles of racemic α-hydroxy ketones 4e2 plotted versus time within 9 h. | |
 |
| | Scheme 4 A plausible asymmetric transfer catalytic pathway of unsymmetrical benzil 3e2. | |
Conclusions
This work presented the successful reductive transformation of unsymmetrical benzils to optically active syn-(S,S)-1,2-diols in terms of good activity and occasionally excellent diastereoselectivity and enantioselectivity. Through dynamic kinetic study on the entire catalytic process monitored using chiral HPLC, a plausible catalytic hydrogenation pathway of unsymmetrical benzil to hydrobenzoin was proposed.
Experimental section
General remarks
All chemicals were purchased and used without any further purification. Unsymmetrical benzils 3a–f and (R,R)-Ru catalysts a, b, and c were synthesized according to the reference, and were ascertained by 1H and 13C NMR.8
Instrumentation and methods
TLC, where applicable, was performed on pre-coated aluminium-backed plates and spots were made visible by quenching ultraviolet (UV) fluorescence (λ = 254 nm). Mass spectra (MS) performed on mass spectrometer (Brucker Daltonics, USA, Brucker Co.) with HCT ultra ion trap. 1H NMR were performed on a Bruker AV-300 NMR instrument at 300.1 MHz. All chemical shifts were reported downfield in ppm relative to hydrogen resonance of TMS. The anti/syn ratios of 1,2-diols 6a–f were determined by 1H NMR of crude products in CDCl3. For example, the chemical shifts of CHOH in 6e2 and 7e2 were 4.65 and 4.78 ppm, respectively. The enantiomeric excesses (%ee) of products with two pairs of enantiomers were determined on Agilent LC-1200 HPLC with a Lux 5u Amylose-2 or Daicel Chiralcel OJ-H chiral column: eluents, (n-hexane–2-propanol = 80/20); 20 °C; UV detector, 254 nm; flow rate, 0.5 mL min−1. Process monitoring containing a mixture of 3e2 (tR = 39.945 min), 4e2 [tR = 54.111 min (S) and 68.884 min (R)], 5e2 [tR = 65.063 min (S) and 76.590 min (R)], 6e2 [tR = 47.257 min (R,R) and 57.126 min (S,S)] and 7e2 [tR = 61.603 min (R,S) and 62.851 min (S,R)] was completely isolated and identified using HPLC on an OJ-H column: eluents, n-hexane–i-PrOH (90:10); flow rate, 1.0 mL min−1; UV detector, λ = 220 (see Fig. S1 in ESI†).
General procedure for 1,3-diketones 2a–f
Sodium hydride (60% in mineral oil, 1.0 g, 25 mmol) was added to a 100 mL three-necked, round-bottom flask. This solution was flushed thrice with Ar atmosphere and cooled to 0 °C to 5 °C. Subsequently, the charged anhydrous THF (35 mL) and a mixture of 4-methoxyacetophenone (1.5 g, 10 mmol) and ethyl benzoate (0.15 g, 1.0 mmol) were added. After stirring for 20 min, another aliquot of ethyl benzoate (1.5 g, 10 mmol) was added to the reaction mixture. The mixture was refluxed for 16 h and cooled to room temperature. Subsequently, ethyl acetate and 10% diluted HCl were added to dissolve all kinds of solids. The organic phase was separated and washed thrice with brine (50 mL × 3) and dried on anhydrous magnesium sulfate. After removal of organic solvent, the residue was purified via chromatography eluting with petroleum ether–ethyl acetate (v/v = 80/1) to produce the corresponding 1,3-diketones 2a–f.
Typical procedure for unsymmetric benzils 3a–f
tert-Butyl nitrite (4.5 mL, 2.5 mmol) was added to a mixture of FeCl3 (16 mg, 20 mol%) and 1,3-diketone (0.5 mmol). The reaction mixture was stirred at 30 °C under air for 12 h. After the reaction was completed, ethyl acetate (2 mL) was added, and the solids were filtered through a Celite pad. After the solvent was evaporated under reduced pressure, the residue was subjected to flash column chromatography by eluting with petroleum ether–ethyl acetate (v/v = 80/1) to obtain the unsymmetric benzils 3a–f.
General procedure for transfer hydrogenation of benzils
A 30 mL tube was filled with benzil (0.5 mmol), (R,R)-Ru catalyst a (3.3 mg, 5.0 × 10−3 mmol), formic acid (1.1 mL, 27.2 mmol), and triethylamine (1.9 mL, 13.6 mmol), successively. The mixture was stirred at 40 °C for 24 h, and 2 mL of ethyl acetate was added. The mixture was evaporated under reduced pressure. The residue was purified via flash column chromatography by eluting with petroleum ether–ethyl acetate (v/v = 3/1) to obtain enantiomerically pure (S,S)-hydrobenzoins 6a–f.
6a1 + 7a1. 20.4%, white solid. δH (300 MHz, CDCl3, Me4Si) 7.36–7.27 (m, 1H, Ar-H), 7.25–7.10 (m, 5H, Ar-H), 7.03 (d, J = 6.7 Hz, 1H, Ar-H), 6.93–6.79 (m, 2H, Ar-H), [5.14 (d, J = 5.6 Hz), 4.96 (d, J = 5.6 Hz), 4.92 (d, J = 6.5 Hz), 4.86 (d, J = 6.5 Hz)] (2H, CHOH). Anal. calcd for C14H13FO2: C, 72.40; H, 5.64; O, 13.78. Found: C, 72.36; H, 5.53; O, 13.72%. Mass (MS): m/z 233.1 [M + H]+. Phenomenex Lux 5u Amylose-2 chiral column: (S,S)-6a1, tR = 52.35 min; (R,R)-6a1, tR = 32.69 min; (R,S or S,R)-7a1, tR = 41.15, 45.17 min.
6a2. 70.2%, white solid. [α]25D = −56.2 (c 0.86, CDCl3). δH (300 MHz, CDCl3, Me4Si) 7.25–7.21 (m, 3H, Ar-H), 7.19–7.06 (m, 3H, Ar-H), 6.99–6.87 (m, 2H, Ar-H), 6.81 (d, J = 7.7 Hz, 1H, Ar-H), [4.85–4.82 (m) and 4.80–4.62 (m)] (2H, CHOH), 2.67 (s, 2H, OH). δC (Me4Si) 160.2, 138.4, 134.0, 128.8, 127.6, 127.1, 126.4, 115.3 (Ph), 79.8, 78.2 (CH). Anal. calcd for C14H13FO2: C, 72.40; H, 5.64; O, 13.78. Found: C, 72.34; H, 5.57; O, 13.79%. Mass (MS): m/z 233.1 [M + H]+. Daicel Chiralcel OJ-H column: (S,S)-6a2, tR = 25.36 min (major); (R,R)-6a2, tR = 37.85 min; (R,S or S,R)-7a2, tR = 31.70, 33.55 min.
6a3. 91.8%, white solid. δH (300 MHz, CDCl3, Me4Si) 7.26–7.22 (m, 3H, Ar-H), 7.12–7.02 (m, 4H, Ar-H), 6.89 (t, J = 8.7 Hz, 2H, Ar-H), [4.83–4.79 (m), 4.69–4.59 (m)] (2H, CHOH), 2.70 (s, 2H, OH). δC (Me4Si) 162.6, 143.1, 138.3, 130.6, 129.0, 127.6, 127.0, 122.8, 114.4, 114.0 (Ph), 79.6, 77.8 (CH). Anal. calcd for C14H13FO2: C, 72.40; H, 5.64; O, 13.78. Found: C, 72.39; H, 5.63; O, 13.76%. Mass (MS): m/z 233.1 [M + H]+. Phenomenex Lux 5u Amylose-2 chiral column: (S,S)-6a3, tR = 34.04 min (major); (R,R)-6a3, tR = 31.65 min; (R,S or S,R)-7a3, tR = 20.96, 26.94 min.
6b1 + 7b1. 22.6%, white solid. δH (300 MHz, CDCl3, Me4Si) 7.62 (d, J = 6.5 Hz, 1H, Ar-H), 7.33–7.27 (m, 4H, Ar-H), 7.25–7.18 (m, 2H, Ar-H), 7.15–7.11 (m, 2H, Ar-H), [5.48 (d, J = 4.7 Hz), 5.26 (d, J = 4.5 Hz), 5.05 (d, J = 4.7 Hz), 4.95 (d, J = 4.5 Hz)] (2H, CHOH), 2.66 (s, 2H, OH). Anal. calcd for C14H13ClO2: C, 67.61; H, 5.27; O, 12.87. Found: C, 67.59; H, 5.23; O, 12.79%. Mass (MS): m/z 249.1 [M + H]+. Phenomenex Lux 5u Amylose-2 chiral column: (S,S)-6b1, tR = 34.62 min (major); (R,R)-6b1, tR = 27.05 min; (R,S or S,R)-7b1, tR = 23.39, 24.99 min.
6b2. 95.9%, white solid. δH (300 MHz, CDCl3, Me4Si) 7.33–7.27 (m, 1H, Ar-H), 7.26–7.22 (m, 3H, Ar-H), 7.19 (s, 1H, Ar-H), 7.14–7.10 (m, 3H, Ar-H), 6.90 (d, J = 7.5 Hz, 1H, Ar-H), [4.84–4.79 (m), 4.74–4.60 (m)] (2H, CHOH), 2.87 (s, 2H, OH). δC (Me4Si) 142.8, 138.8, 134.1, 132.3, 128.6, 127.9, 127.4, 127.1, 125.7, 124.8 (Ph), 80.0, 79.4 (CH). Anal. calcd for C14H13ClO2: C, 67.61; H, 5.27; O, 12.87. Found: C, 67.55; H, 5.20; O, 12.72%. Mass (MS): m/z 249.1 [M + H]+.
6b3. 97.1%, white solid. [α]25D = −63.0 (c 0.92, CDCl3). δH (300 MHz, CDCl3, Me4Si) 7.26–7.24 (m, 3H, Ar-H), 7.21 (s, 1H, Ar-H), 7.18 (s, 1H, Ar-H), 7.17–7.09 (m, 2H, Ar-H), 7.03 (d, J = 8.4 Hz, 2H, Ar-H), [4.83 (s), 4.66 (dd, J = 18.9, 7.5 Hz)] (2H, CHOH), 2.34 (s, 2H, OH). δC (Me4Si) 138.9, 137.1, 133.5, 129.7, 129.0, 128.4, 127.1, 126.4 (Ph), 80.0, 79.7 (CH). Anal. calcd for C14H13ClO2: C, 67.61; H, 5.27; O, 12.87. Found: C, 67.55; H, 5.20; O, 12.72%. Mass (MS): m/z 249.1 [M + H]+. Phenomenex Lux 5u Amylose-2 chiral column: (S,S)-6b3, tR = 40.39 min (major); (R,R)-6b3, tR = 22.30 min; (R,S or S,R)-7b1, tR = 35.31, 38.89 min.
6c1. 55.4%, white solid. [α]25D = −69.3 (c 0.84, CDCl3). δH (300 MHz, CDCl3, Me4Si) 7.58–7.47 (m, 6H, Ar-H), 7.12–7.06 (m, 3H, Ar-H), [4.82 (s), 4.69 (s)] (2H, CHOH), 3.04 (s, 2H, OH). δC (Me4Si) 144.6, 138.4, 133.4, 130.2, 128.9, 127.9, 127.7, 127.1, 126.5, 120.3 (Ph), 79.8, 76.0 (CH). Anal. calcd for C14H13BrO2: C, 57.36; H, 4.47; O, 10.92. Found: C, 57.35; H, 4.40; O, 10.87%. Mass (MS): m/z 293.0 [M + H]+. Phenomenex Lux 5u Amylose-2 chiral column: (S,S)-6c1, tR = 27.80 min (major); (R,R)-6c1, tR = 31.90 min; (R,S and S,R)-7c1, tR = 39.18 min.
6c2. 97.6%, white solid. δH (300 MHz, CDCl3, Me4Si) 7.36 (d, J = 6.4 Hz, 2H, Ar-H), 7.30–7.27 (m, 1H, Ar-H), 7.25–7.22 (m, 2H, Ar-H), 7.13–7.04 (m, 3H, Ar-H), 6.94 (d, J = 7.7 Hz, 1H, Ar-H), [4.81 (m), 4.71–4.62 (m)] (2H, CHOH), 2.37 (s, 2H, OH). δC (Me4Si) 143.2, 138.6, 132.2, 130.8, 129.8, 129.0, 127.9, 127.3, 126.4, 123.8 (Ph), 80.1, 79.2 (CH). Anal. calcd for C14H13BrO2: C, 57.36; H, 4.47; O, 10.92. Found: C, 57.31; H, 4.42; O, 10.82%. Mass (MS): m/z 293.0 [M + H]+.
6c3. 93.2%, white solid. [α]25D = −65.8 (c 1.02, CDCl3). δH (300 MHz, CDCl3, Me4Si) 7.34 (d, J = 8.4 Hz, 2H, Ar-H), 7.25 (d, J = 3.2 Hz, 2H, Ar-H), 7.12–7.07 (m, 2H, Ar-H), 6.97 (d, J = 8.4 Hz, 2H, Ar-H), [4.82 (s), 4.65 (q, J = 7.5 Hz)] (2H, CHOH), 2.91 (s, 2H, OH). δC (Me4Si) 138.9, 137.8, 132.2, 129.0, 127.6, 127.1, 126.5, 122.0 (Ph), 80.1, 80.0 (CH3). Anal. calcd for C14H13BrO2: C, 57.36; H, 4.47; O, 10.92. Found: C, 57.31; H, 4.43; O, 10.86%. Mass (MS): m/z 293.0 [M + H]+. Phenomenex Lux 5u Amylose-2 chiral column: (S,S)-6c3, tR = 43.17 min (major); (R,R)-6c3, tR = 23.23 min; (R,S and S,R)-7c3, tR = 35.72, 40.22 min.
6d1 + 7d1. 22.7%, white solid. δH (300 MHz, CDCl3, Me4Si) 8.12–8.04 (m, 1H, Ar-H), 7.64–7.55 (m, 1H, Ar-H), 7.50–7.45 (m, 1H, Ar-H), 7.36–7.28 (m, 1H, Ar-H), 7.26–7.19 (m, 2H, Ar-H), 7.15–7.09 (m, 2H, Ar-H), 6.95 (d, J = 7.5 Hz, 1H, Ar-H), [5.13 (d, J = 5.9 Hz), 4.93 (d, J = 7.4 Hz), 4.86 (d, J = 5.9 Hz), 4.76 (d, J = 7.4 Hz)] (2H, CHOH), 3.57 (s, 2H, OH), [2.65 (s), 1.75 (s)] (3H, CH3). Anal. calcd for C15H16O2: C, 78.92; H, 7.06; O, 14.02. Found: C, 78.84; H, 6.95; O, 13.96%. Mass (MS): m/z 229.1 [M + H]+. Daicel Chiralcel OJ-H column: (S,S)-6d1, tR = 20.94 min (major); (R,R)-6d1, tR = 25.56 min; (R,S and S,R)-7d1, tR = 33.03 min.
6d2. 78.7%, white solid. δH (300 MHz, CDCl3, Me4Si) 7.24 (d, J = 2.2 Hz, 3H, Ar-H), 7.16–7.09 (m, 3H, Ar-H), 7.04 (d, J = 7.6 Hz, 1H, Ar-H), 6.99 (s, 1H, Ar-H), 6.90 (d, J = 7.3 Hz, 1H, Ar-H), [4.78 (q, J = 6.4 Hz), 4.70 (q, J = 7.0 Hz)] (2H, CHOH), [2.32 (s), 2.28 (s)] (3H, CH3). δC (Me4Si) 140.8, 139.6, 138.0, 129.5, 129.0, 128.8, 127.7, 127.2, 126.8, 124.0 (Ph), 80.3, 79.8 (CH), 21.6 (CH3). Anal. calcd for C15H16O2: C, 78.92; H, 7.06; O, 14.02. Found: C, 78.84; H, 6.97; O, 13.92%. Mass (MS): m/z 229.1 [M + H]+. Daicel Chiralcel OJ-H column: (S,S)-6d2, tR = 28.07 min (major); (R,R)-6d2, tR = 25.56 min; (R,S and S,R)-7d2, tR = 30.93, 33.20 min.
6d3. 80.6%, white solid. δH (300 MHz, CDCl3, Me4Si) 7.22–7.20 (m, 3H, Ar-H), 7.12–7.10 (m, 2H, Ar-H), 7.04–6.98 (m, 4H, Ar-H), [4.68 (q, J = 6.2 Hz), 4.65 (q, J = 7.4 Hz)] (2H, CHOH), 3.31 (s, 2H, OH), [2.40 (s), 2.29 (s)] (3H, CH3). δC (Me4Si) 138.0, 137.1, 134.8, 129.8, 128.6, 128.0, 127.2, 125.5 (Ph), 80.2, 80.0 (CH), 21.3 (CH3). Anal. calcd for C15H16O2: C, 78.92; H, 7.06; O, 14.02. Found: C, 78.80; H, 7.93; O, 13.94%. Mass (MS): m/z 229.1 [M + H]+. Phenomenex Lux 5u Amylose-2 chiral column: (S,S)-6d3, tR = 54.11 min (major); (R,R)-6d3, tR = 51.10 min; (R,S and S,R)-7d3, tR = 28.84, 44.54 min.
6e1 + 7e1. 31.2%, white solid. δH (300 MHz, CDCl3, Me4Si) 7.27 (s, 1H, Ar-H), 7.25–7.24 (m, 1H, Ar-H), 7.22–7.16 (m, 3H, Ar-H), 7.15–7.12 (m, 1H, Ar-H), 6.92–6.82 (m, 3H, Ar-H), [5.17 (d, J = 5.5 Hz), 4.94 (d, J = 5.5 Hz), 4.86 (q, J = 6.6 Hz)] (2H, CHOH), [3.76 (s), 3.71 (s)] (3H, OCH3), 2.69 (s, 2H, OH). Anal. calcd for C15H16O3: C, 73.75; H, 6.60; O, 19.65. Found: C, 73.69; H, 6.53; O, 19.34%. Mass (MS): m/z 245.1 [M + H]+. Phenomenex Lux 5u Amylose-2 chiral column: (S,S)-6e1, tR = 68.05 min; (R,R)-6e1, tR = 46.31 min; (R,S and S,R)-7e1, tR = 52.27, 60.47 min.
6e2. 93.5%, white solid. [α]25D = −113.6 (c 1.10, CDCl3). δH (300 MHz, CDCl3, Me4Si) 7.25–7.21 (m, 3H, Ar-H), 7.12 (t, J = 7.6 Hz, 3H, Ar-H), 6.75 (dd, J = 8.2, 1.9 Hz, 1H, Ar-H), 6.67 (d, J = 9.2 Hz, 2H, Ar-H), [4.78 (s), 4.65 (s)] (2H, CHOH), 3.68 (s, 3H, OCH3), 2.88 (s, 2H, OH). δC (Me4Si) 160.8, 141.8, 138.4, 129.9, 128.9, 127.6, 127.1, 119.4, 113.2, 111.2 (Ph), 80.3, 80.0 (CH), 55.8 (OCH3). Anal. calcd for C15H16O3: C, 73.75; H, 6.60; O, 19.65. Found: C, 73.71; H, 6.55; O, 19.49%. Mass (MS): m/z 245.1 [M + H]+. Daicel Chiralcel OJ-H column: (S,S)-6e1, tR = 53.65 min (major); (R,R)-6e1, tR = 46.31 min; (R,S and S,R)-7e1, tR = 59.21, 61.10 min.
6e3. 68.5%, white solid. δH (300 MHz, CDCl3, Me4Si) 7.26–7.21 (m, 3H, Ar-H), 7.58–7.47 (m, 2H, Ar-H), 7.03 (d, J = 8.6 Hz, 2H, Ar-H), 6.75 (d, J = 8.6 Hz, 2H, Ar-H), [4.78 (q, J = 6.1 Hz), 4.65 (q, J = 7.7 Hz)] (2H, CHOH), 3.76 (s) (3H, OCH3), 2.98 (s, 2H, OH). δC (Me4Si) 159.8, 138.0, 131.1, 128.3, 128.1, 127.8, 127.3, 127.0 (Ph), 80.5, 80.0 (CH), 55.5 (OCH3). Anal. calcd for C15H16O3: C, 73.75; H, 6.60; O, 19.65. Found: C, 73.65; H, 6.49; O, 19.52%. Mass (MS): m/z 245.1 [M + H]+.
6f2. 92.2%, white solid. [α]25D = −89.8 (c 0.96, CDCl3). δH (300 MHz, CDCl3, Me4Si) 7.46 (d, J = 7.7 Hz, 1H, Ar-H), 7.30 (d, J = 7.9 Hz, 2H, Ar-H), 7.25–7.17 (m, 4H, Ar-H), 7.05–7.03 (m, 2H, Ar-H), [4.88 (d, J = 5.1 Hz), 4.82 (d, J = 5.3 Hz), 4.71 (d, J = 7.4 Hz), 4.57 (d, J = 7.4 Hz)] (2H, CHOH), 3.20 (s, 2H, OH). δC (Me4Si) 141.2, 138.8, 131.3, 129.8, 128.9, 127.9, 127.3, 125.1, 124.2 (Ph), 123.4 (CF3, q, 2JC–F = 15.6 Hz), 80.3, 80.0 (CH). Anal. calcd for C15H13F3O2: C, 63.83; H, 4.64; O, 11.34. Found: C, 63.74; H, 4.56; O, 11.29%. Mass (MS): m/z 283.1 [M + H]+. Phenomenex Lux 5u Amylose-2 chiral column: (S,S)-6f2, tR = 20.79 min (major); (R,R)-6f2, tR = 14.15 min; (R,S and S,R)-7f2, tR = 16.64, 19.67 min.
6f3. 94.9%, white solid. [α]25D = −87.1 (c 1.02, CDCl3). δH (300 MHz, CDCl3, Me4Si) 7.45 (d, J = 8.1 Hz, 2H, Ar-H), 7.24 (d, J = 3.4 Hz, 2H, Ar-H), 7.16 (d, J = 8.0 Hz, 2H, Ar-H), 7.07 (dd, J = 6.4, 2.8 Hz, 2H, Ar-H), [4.88 (d, J = 5.4 Hz), 4.82 (d, J = 5.3 Hz), 4.72 (d, J = 7.4 Hz), 4.60 (d, J = 7.4 Hz)] (2H, CHOH), 3.03 (s, 2H, OH). δC (Me4Si) 141.9, 138.8, 130.2, 128.5, 127.6, 127.0, 126.1, 125.4 (Ph), 123.8 (CF3, q, 2JC–F = 18,3 Hz), 80.2, 80.0 (CH). Anal. calcd for C15H13F3O2: C, 63.83; H, 4.64; O, 11.34. Found: C, 63.74; H, 4.56; O, 11.29%. Mass (MS): m/z 283.1 [M + H]+. Phenomenex Lux 5u Amylose-2 chiral column: (S,S)-6f3, tR = 21.61 min (major); (R,R)-6f3, tR = 15.35 min; (R,S and S,R)-7f3, tR = 17.85, 19.81 min.
Acknowledgements
The work was financially supported by the National Science Foundation of China (grants 21071116) and Chongqing Scientific Foundation (CSTC, 2010BB4126).
Notes and references
-
(a) L. Bagnoli, C. Scarponi, M. G. Rossi, L. Testaferri and M. Tiecco, Chem. – Eur. J., 2011, 17, 993–999 CrossRef CAS PubMed;
(b) C. Ramarao, R. Nandipati, R. Navakoti and R. Kottamasu, Tetrahedron Lett., 2012, 53, 637–640 CrossRef CAS PubMed;
(c) L. X. Wang, X. Y. Wang, J. N. Cui, W. M. Ren, N. Meng, J. Y. Wang and X. H. Qian, Tetrahedron: Asymmetry, 2010, 21, 825–830 CrossRef CAS PubMed;
(d) C. Zhu, Y. Shi, M. H. Xu and G. Q. Lin, Org. Lett., 2008, 10, 1243–1246 CrossRef CAS PubMed;
(e) E. Wojaczynska and J. Skarzewski, Tetrahedron: Asymmetry, 2008, 19, 593–597 CrossRef CAS PubMed;
(f) D. Nakamura, K. Kakiuchi, K. Koga and R. Shirai, Org. Lett., 2006, 8, 6139–6142 CrossRef CAS PubMed;
(g) R. Pedrosa, S. Sayalero, M. Vicente and A. Maestro, J. Org. Chem., 2006, 71, 2177–2180 CrossRef CAS PubMed;
(h) R. S. Reddy, I. N. C. Kirana and A. Sudalai, Org. Biomol. Chem., 2012, 10, 3655–3661 RSC;
(i) K. Akshay, S. Sarbjit, K. Vikas and S. C. Swapandeep, Org. Biomol. Chem., 2011, 9, 2731–2742 RSC.
-
(a) M. Tanaka, Y. Demizu, M. Nagano, M. Hama, Y. Yoshida, M. Kurihara and H. Suemune, J. Org. Chem., 2007, 72, 7750–7756 CrossRef CAS PubMed;
(b) H. K. Chenault, L. F. Chafin and S. Liehr, J. Org. Chem., 1998, 63, 4039–4045 CrossRef CAS;
(c) C. B. Shinisha and R. B. Sunoj, Org. Lett., 2009, 11, 3242–3245 CrossRef CAS PubMed;
(d) P. Molnar, P. Thorey, G. Bansaghi, E. Szekely, L. Poppe, A. Tomin, S. Kemeny, E. Fogassy and B. Simandi, Tetrahedron: Asymmetry, 2008, 19, 1587–1592 CrossRef CAS PubMed;
(e) K. Justyna, C. S. Robert, A. S. Christopher, M. H. Syed, H. Matthias, L. Steffen, Z. Kirsten, K. Wolfgang, P. Martina and R. Dörte, Catal. Sci. Technol., 2012, 2, 1580–1589 RSC.
-
(a) N. H. Taniguchi and M. T. Uemura, Angew. Chem., Int. Ed., 1999, 38, 1232–1235 CrossRef CAS;
(b) Y. H. Zhao, R. L. Ma, X. W. Sun and G. Q. Lin, Synthesis, 2012, 2763–2769 CAS;
(c) M. Kitamura, K. Shiomi, D. Kitahara, Y. Yamamoto and T. Okauchi, Synlett, 2010, 1359–1362 CrossRef CAS PubMed;
(d) J. T. Sun, Z. Y. Dai, C. S. Li, X. Pan and C. J. Zhu, J. Organomet. Chem., 2009, 694, 3219–3221 CrossRef CAS PubMed;
(e) E. V. Sergeeva, V. I. Rozenberg, D. Y. Antonov, E. V. Vorontsov, Z. A. Starikova, I. V. Fedyanin and H. Hopf, Chem. – Eur. J., 2011, 11, 6944–6961 CrossRef PubMed.
-
(a) D. R. Boyd, N. D. Sharma, M. V. Berberian, K. S. Dunne, C. Hardacre, M. Kaik, B. Kelly, J. F. Malone, S. T. McGregor and P. J. Stevenson, Adv. Synth. Catal., 2010, 352, 855–868 CrossRef CAS;
(b) P. Jiao, M. Kawasaki and H. Yamamoto, Angew. Chem., Int. Ed., 2009, 48, 3333–3336 CrossRef CAS PubMed;
(c) E. Vedrenne, O. A. Wallner, M. Vitale, F. Schmidt and V. K. Aggarwal, Org. Lett., 2009, 11, 165–168 CrossRef CAS PubMed;
(d) L. Ma, D. M. Zhao, L. Chen, X. Wang, Y. L. Chen and J. K. Shen, Tetrahedron, 2012, 68, 8704–8711 CrossRef CAS PubMed.
-
(a) S. B. Han, H. Han and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 1760–1761 CrossRef CAS PubMed;
(b) A. Carlone, G. Bartoli, M. Bosco, F. Pesciaioli, P. Ricci, L. Sambri and P. Melchiorre, Eur. J. Org. Chem., 2007, 5492–5495 CrossRef CAS;
(c) T. Koike, K. Murata and T. Ikariya, Org. Lett., 2000, 2, 3833–3836 CrossRef CAS PubMed;
(d) K. Murata, K. Okano, M. Miyagi, H. Iwane, R. Noyori and T. Ikariya, Org. Lett., 1999, 1, 1119–1121 CrossRef CAS.
-
(a) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS;
(b) J. X. Gao, T. Ikariya and R. Noyori, Organometallics, 1996, 15, 1087–1089 CrossRef CAS;
(c) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS;
(d) N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 4916–4917 CrossRef CAS;
(e) X. Xu, R. Wang, J. Wan, X. Ma and J. Peng, RSC Adv., 2013, 3, 6747–6751 RSC;
(f) R. Wang, J. Wan, X. Ma, X. Xu and L. Liu, Dalton Trans., 2013, 42, 6513–6522 RSC.
- L. H. Huang, K. Cheng, B. B. Yao, Y. J. Xie and Y. H. Zhang, J. Org. Chem., 2011, 76, 5732–5737 CrossRef CAS PubMed.
- M. C. Fragnelli, P. Hoyos, D. Romano, R. Gandolfi, A. R. Alcántara and F. Molinari, Tetrahedron, 2012, 68, 523–528 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR of 2a–f, 3a–f, 4e2, 5e2, 6a–f; HPLC diagram of 6a–f; kinetic data of 3e2, 4e2 and 5e2 in catalytic process. See DOI: 10.1039/c3ra47129b |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.