Interactions of anti-proliferative and anti-platelet drugs with self-assembled monolayers: a future strategy in stent development
Abdolreza Mirmohseni*a,
Javad Hosseinia,
Maryam Shojaeib and
Soodabeh Davarancd
aDepartment of Applied Chemistry, Faculty of Chemistry, University of Tabriz, 5166616471, Tabriz, Iran. E-mail: mirmohseni@tabrizu.ac.ir; Fax: +98 411 334 0191; Tel: +98 411 339 3171
bAnimal Biology Department, Faculty of Natural Sciences, University of Tabriz, 5166616471, Tabriz, Iran
cDrug Applied Research Center, Tabriz University of Medical Sciences, 5166414766, Tabriz, Iran
dMedicinal Nanotechnology Department, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences, 5166414766, Tabriz, Iran
First published on 5th December 2013
Abstract
The current study is part of an overall goal to develop new drug-eluting stents (DES). In this paper, for the first time, simultaneous elution of anti-proliferative and anti-platelet drugs from self-assembled monolayers was investigated. Methyl- and carboxyl-terminated mixed self-assembled monolayers were prepared on gold (Au) surfaces. The samples were characterized by X-ray photoelectron spectroscopy (XPS), atomic force microscopy (AFM), contact angle goniometry (CA), Fourier transform infrared spectroscopy (FTIR), and scanning electron microscopy (SEM). To demonstrate that the resulting structures can be used for simultaneous delivery of paclitaxel and dipyridamole, the drug release in phosphate-buffered saline (PBS) was studied using high performance liquid chromatography (HPLC). Obtained results confirmed successful transfer of drugs to PBS. Released dipyridamole levels were significantly higher (p < 0.05) than the delivered paclitaxel after 1, 2, 3, 7, 14 and 28 days. These findings indicate that the proposed method might be suitable for development of an advanced generation of drug-eluting stents.
Introduction
Coronary arteries supply the cardiac muscles with nutrients and oxygen. Coronary artery disease (CAD) occurs when the coronary arteries become narrowed or blocked by deposition of atheromatous plaques. Millions of people die each year from CAD and it is still the leading cause of death worldwide.1,2 A number of therapeutic interventions such as angioplasty, angiogenesis, brachytherapy and bypass grafting have been developed to restore normal blood flow.3 Among them, angioplasty is an increasingly important common invasive procedure for treatment of cardiovascular disorders. Percutaneous transluminal coronary angioplasty (PTCA) or balloon angioplasty is widely used to reopen blocked arteries. However, restenosis in 30% to 60% of patients remains an unresolved problem. Elastic recoil and negative remodeling which play a crucial role in restenosis can be reduced using stent implantation. In-stent restenosis rate is reported to be 20–30%.4–6 The key problem with the bare metal stents is that they can cause neointimal hyperplasia (NH). NH is characterized by the recruitment and proliferation of smooth muscle cells and fibroblasts. Anti-proliferative drugs like paclitaxel prevent NH by inhibiting the growth of smooth muscle cells.6–8 Systemic drug delivery can lead to system toxicity. Hence, in recent years, researchers have shown interest in local drug delivery methods.3,9 The introduction of drug-eluting stents (DES) has reduced the restenosis rates to less than 5%.6 DES that deliver anti-proliferative drugs locally to decrease the risk of in-stent restenosis are widely implanted.10,11 Despite their clinical success, DES have two problems in use. First, polymers are used in most commercially available DES to control drug release kinetics. The use of polymers as drug carriers can result in late/very late stent thrombosis (LST/VLST) and can lead to hypersensitivity reactions.12–14 Additionally, the occurrence of mechanical defects or irregularities in the polymer layers is possible.15 Second, metal stents in coronary arteries may serve as a nidus for aggregation of platelets.16 Anti-platelet drugs are increasingly being prescribed to reduce platelet aggregation. However, systemically administrated drugs may not have been delivered in sufficient concentrations to target sites. Another problem with systemic drug delivery is that it can cause adverse effects. Furthermore, interruption of systemic anti-platelet therapy is considered to be an important risk factor for myocardial infarction after stent implantation.17,18The term “self-assembly” has come to be used to refer to a process in which disordered molecules form an ordered system in absence of external direction.19 Many studies have attempted to utilize this strategy in therapeutic applications.20–23 Recently, several research groups have attempted to explain the use of self-assembled monolayers in nonpolymer-based drug delivery platforms. In previous publications metal surfaces (Ti, Co–Cr alloy, etc.) have been coated with carboxylic acid or hydroxyl terminated monolayers and therapeutic agents (flufenamic acid, paclitaxel, etc.) have been attached to self-assembled monolayers.9,24,25 The findings are valuable for designing new stents. However, in reviewing the literature no research was found on the investigation of drug elution from mixed self-assembled monolayers that have two components. The main limitation of single-component monolayers is the presence of undesirable interactions between neighbouring terminal groups prohibits the favourable attachment of therapeutic agent to self-assembled monolayers. In addition to the above mentioned knowledge gap, up to now the simultaneous release of anti-proliferative and anti-platelet drugs has been neglected. We reported earlier using mixed self-assembled monolayers to prepare polymer-free DES as a solution for the first problem (late/very late stent thrombosis).26 Our concern now is to find out a way to solve the second problem (platelet aggregation). It would be interesting to study if it is possible to use simultaneous local delivery of anti-proliferative and anti-platelet drugs as a new strategy for DES development. The aim of this paper was to study simultaneous delivery of dipyridamole (as a platelet inhibitor) and paclitaxel (as an anti-proliferative agent) from gold (Au) surfaces coated with 12-mercaptododecanoic acid and 1-nonanethiol mixed self-assembled monolayers. Fig. 1 shows a schematic view of our proposed drug-eluting system and its implantation. Principally gastrointestinal disturbance and bleeding are two side effects of aspirin. These problems and aspirin modes efficacy have heightened the need for additional or alternative anti-platelet drugs.27 Dipyridamole which inhibits the uptake of adenosine into blood cells prevents platelet aggregation.28 On the other hand, paclitaxel (Taxol) which has been isolated from the inner bark of Pacific yew tree is an anti-proliferative drug. Paclitaxel binds to β-tublin of microtubules and inhibits cellular replication.29 Recently, researchers have reported that biological effect of paclitaxel increases in the presence of dipyridamole. Also, it is proven that compared with the sum of paclitaxel and dipyridamole biological effects when they act alone, the effects of the dipyridamole/paclitaxel combination is greater. Simultaneous delivery of the above mentioned drugs could be developed for use in many medical devices such as intravascular stents, intravascular infusion catheters, drug delivery catheters, anastomotic connector devices, breast implants, lip implants, hemodialysis access devices and nasal implants.30
 | ||
| Fig. 1 A schematic view of proposed drug-eluting stent system and its implantation. | ||
Experimental
Materials
Acetonitrile, dipyridamole, 12-mercaptododecanoic acid (HS(CH2)10CO2H), methanol, 1-nonanethiol (CH3(CH2)8SH) and paclitaxel were purchased from Sigma-Aldrich. Absolute ethanol and acetone were obtained from Scharlau (Spain). Phosphate buffered saline was purchased from CMG (Cyto Matin Gene Ltd, Iran). All chemicals were used as received without any further purification. Gold plates were bought from ICM (International Crystal Manufacturer Co, Oklahoma, USA).Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetone (1:1, v/v) for 24 h. Au substrates were then rinsed with distilled water. After rinsing, the specimens were ultrasonically cleaned in acetone, deionized water and 50% ethanol each for 5 min at room temperature. Finally, the samples were rinsed using absolute ethanol.
:acetone (1:1, v/v) for 24 h. Au substrates were then rinsed with distilled water. After rinsing, the specimens were ultrasonically cleaned in acetone, deionized water and 50% ethanol each for 5 min at room temperature. Finally, the samples were rinsed using absolute ethanol.In order to prepare mixed self-assembled monolayers, cleaned samples were immersed in a mixed ethanolic solution of 12-mercaptododecanoic acid (2 mM, as functional thiolate) and 1-nonanethiol (8 mM, as diluting thiolate) at 25 °C for 25 h. The specimens were then taken out and rinsed with absolute ethanol and subsequently with deionized water. Ultimately, the samples were dried under a stream of pure N2 gas.
Analysis techniques
Drug elution study
:20, v/v) was pumped at a flow rate of 1.2 mL min−1. UV detector (Waters 2489) was fixed at 227 nm for paclitaxel and 280 nm for dipyridamole. In the concentration range of 1–1000 ng mL−1, the calibration graphs were linear with correlation coefficients of 0.999 and 0.998 for dipyridamole and paclitaxel, respectively. The regression equations were calculated as Y = 471662X − 5625 for dipyridamole and Y = 356295X − 7830 for paclitaxel.Statical analysis
The results are presented as mean ± standard deviation. In order to determine the significance at p < 0.05 for 95% confidence, one-way ANOVA was used. Data management and analysis were performed with SPSS version 16.0 software.Results and discussion
Gold was chosen as a model due to its favourable features such as good compatibility with cells, chemical inertness and non-toxicity. Also, Au film has been used as a common substrate for several analytical and spectroscopies techniques. Furthermore, thiols form stable self-assembled monolayers on Au films.5,31–33Dipyridamole and paclitaxel contain hydroxyl groups and hence have a potential for derivatization of carboxyl terminated mixed self-assembled monolayers. Researchers have mentioned hydrogen and covalent bonds as the bonds which affect attachment of drugs to self-assembled monolayers. They have also revealed that the esterification of the mercapto carboxylic acid derivatives by the OH groups of drugs is dependent on certain conditions such as high temperature and the presence of catalysts.9,24 However, in order to prevent the degradation of drugs, high temperature or catalysts were not used in this study. Therefore, in the current research the attachment of paclitaxel and dipyridamole to monolayer is related to electrostatic interactions or hydrogen bonds. In fact, covalent bonds do not have a substantial role in the process.
The methyl-terminated alkane thiols may cause minimizing of undesirable interactions between neighboring –COOH moieties of 12-mercaptododecanoic acid molecules.34 However, phase separation is likely to occur in the mixed monolayer when chain length difference between two components is large (Δn > 4).35,36 With considering all aspects, mixed self-assembled monolayer was prepared using 12-mercaptododecanoic acid and 1-nonanethiol.
XPS characterization
High-resolution XPS spectra of S 2p and N 1s are presented in Fig. 2(a) and Fig. 2(b), respectively. Fig. 2(a) shows XPS S 2p spectra before and after the formation of mixed self-assembled monolayer. No XPS peak was found for the bare surface (Au). However, for the mixed self-assembled monolayer (SAM), the S 2p peak at binding energy of 161.9 ± 0.2 eV was observed. This result confirms successful formation of monolayer. As can be seen in Fig. 2(a) SAM, a shoulder is present on the right hand side of window scan. It is difficult to explain this finding, but it might be related to the interaction of sulfur atoms with different gold sites (step and terrace). Fig. 2(b) presents the N 1s region before (SAM) and after (DP–SAM) attachment of therapeutic agents. The peak at 404.7 ± 0.2 eV for the sample containing dipyridamole (C24H40N8*O4) and paclitaxel (C47H51N*O14) was assigned to nitrogen atoms in the molecular structure of drugs. The results indicate successful attachment of therapeutic agents to the mixed self-assembled monolayer.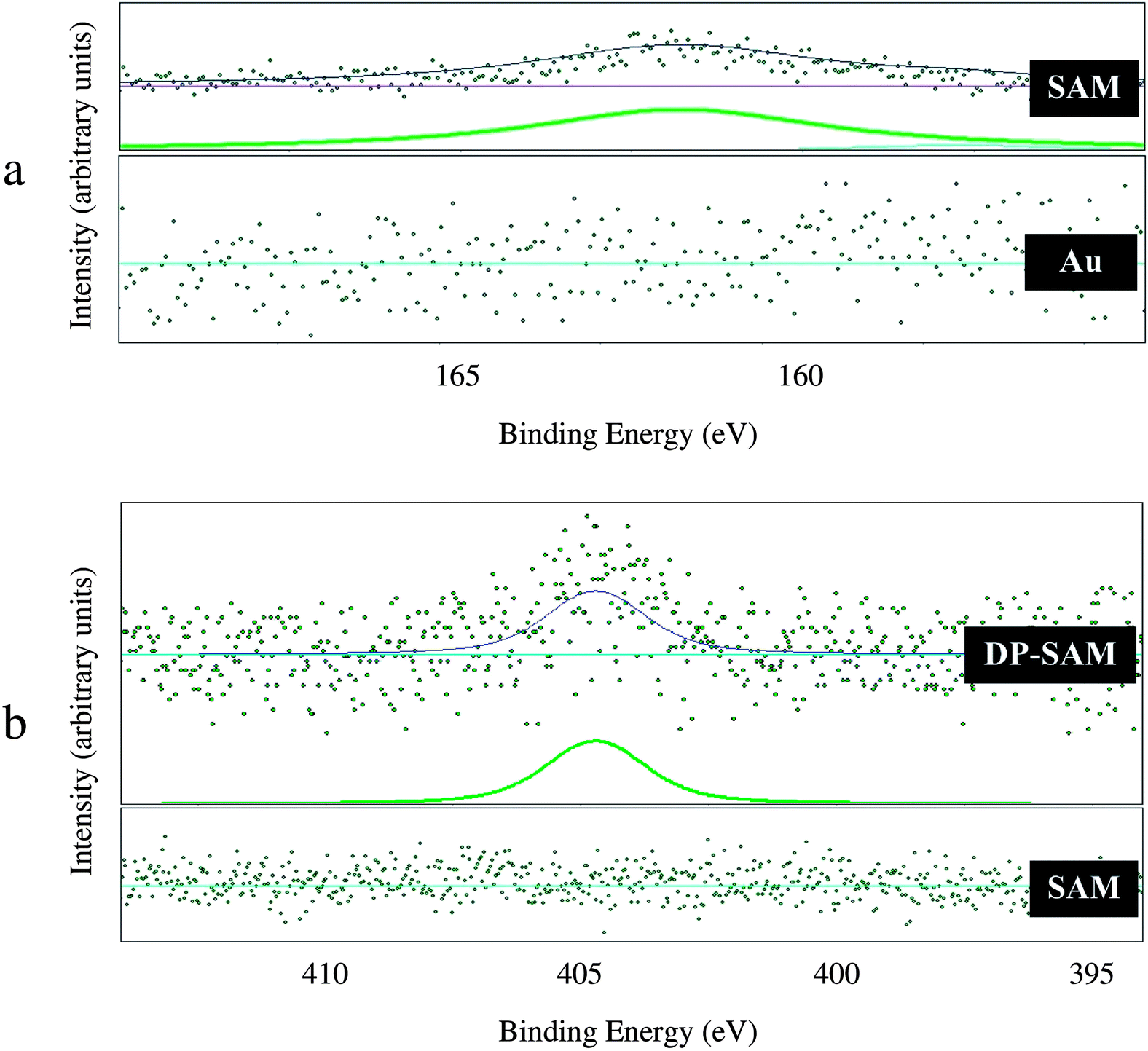 | ||
| Fig. 2 XPS spectra: (a) XPS S 2p spectra of the bare surface (Au) and mixed self-assembled monolayer (SAM) and (b) XPS N 1s spectra of the mixed self-assembled monolayer before (SAM) and after (DP–SAM) the attachment of drugs. | ||
AFM characterization
As it can be seen in the Fig. 3, mixed self-assemble monolayer is well ordered and compact. This result is similar to the findings, which have been reported for homogeneous (single-component) self-assembled monolayers with more than six to seven methylene units. A possible explanation for this might be that linear alkane thiols as the host matrix materials due to their closely packed arrangement form orderly mixed monolayers.35,37 No significant increase in RMS roughness value of the mixed monolayer (15.6 ± 3.0 nm) was observed compared to the uncoated gold surface (14.3 ± 4.1 nm). This also could be related to the formation of a uniform monolayer. | ||
| Fig. 3 AFM images of mixed self-assembled monolayers: (a) contact mode and (b) tapping mode. | ||
SEM characterization
The results obtained from the SEM analysis are presented in Fig. 4. It is apparent from Fig. 4(a) that the surface cleaning procedure employed in the current study is useful for reducing the surface contaminants. As shown in Fig. 4(b), the mixed monolayer film was uniform and dense. From Fig. 4(c) we can see that the uniform structure of the mixed film allowed dipyridamole and paclitaxel molecules to attach to the monolayer end groups. Fig. 4(c) also indicates that there was no agglomeration of therapeutic molecules. | ||
| Fig. 4 SEM images of (a) the bare surface (Au), (b) mixed self-assembled monolayer (SAM) and (c) mixed monolayer after the attachment of dipyridamole and paclitaxel (DP–SAM). | ||
Contact angle measurements
The static contact angle of the monolayer surface was increased from 80 ± 4° to 97 ± 2° after drugs binding. The increase in water contact angle is attributed to a decrease in the number of monolayer hydrophilic moieties (–COOH) caused by the formation of bonds between the –COOH end groups and the hydrophobic drugs.FTIR characterization
Fig. 5 shows the results obtained from the FTIR measurements. Peaks <2918 cm−1 and <2850 cm−1 (Fig. 5(a)) are assigned to methylene asymmetric and symmetric stretches, respectively. This result is consistent with those of other studies11,25,38 and confirm that the mixed self-assembled monolayer is well ordered. A band for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of the mixed self-assembled monolayer appeared at 1710 cm−1 (Fig. 5(b)). This indicates that adjacent –COOH moieties are not involved in the molecular bonds. FTIR was also used to confirm the attachment of dipyridamole and paclitaxel to the gold surface. Fig. 5(c) shows four peaks at 1742, 1719, 1699 and 1648 cm−1. These results that provide additional evidence for the successful attachment of drugs to the mixed self-assembled monolayer can be summarized as follows:
O stretch of the mixed self-assembled monolayer appeared at 1710 cm−1 (Fig. 5(b)). This indicates that adjacent –COOH moieties are not involved in the molecular bonds. FTIR was also used to confirm the attachment of dipyridamole and paclitaxel to the gold surface. Fig. 5(c) shows four peaks at 1742, 1719, 1699 and 1648 cm−1. These results that provide additional evidence for the successful attachment of drugs to the mixed self-assembled monolayer can be summarized as follows:
 | ||
| Fig. 5 FTIR spectra of (a) mixed self-assembled monolayer (SAM: 2917 and 2838 cm−1), (b) mixed self-assembled monolayer (SAM: 1710 cm−1) and (c) mixed self-assembled monolayer after the attachment of dipyridamole and paclitaxel (DP–SAM: 1742, 1719, 1699 and 1648 cm−1). | ||
(I) 1742 cm−1: ester carbonyl groups (paclitaxel)
(II) 1719 cm−1: ester carbonyl group conjugated with the phenyl ring (paclitaxel)
(III) 1699 cm−1: ester carbonyl group involved in hydrogen bonding (paclitaxel)
(IV) 1648 cm−1: CN double bond (dipyridamole).
Drug elution study
Representative HPLC chromatograms of dipyridamole and paclitaxel are shown in Fig. 6. The retention times of dipyridamole and paclitaxel were ∼12.4 min (Fig. 6(a)) and ∼14.4 min (Fig. 6(b)), respectively. Fig. 7 presents in vitro release profiles. As can be seen, the profiles were independent of drug type and had similar biphasic shapes. This finding is important when considering simultaneous delivery of anti-proliferative and anti-platelet drugs. As another result, at the selected times the amount of eluted dipyridamole was significantly higher than the delivered paclitaxel dose. A possible explanation for this might be that because of their flat structures, dipyridamole molecules can be easily attached to and detached from the mixed self-assembled monolayers. An alternative explanation of the higher loading capacity achieved with dipyridamole can be provided attending to the pKa values of the carboxylic group of the mercaptoacid and the polynitrogenated drug which can be considered as a base. Therefore this compound can be retained more properly by the acid groups present on the surface and this fact can explain the higher loading capacity. Finally, due to the weak nature of the electrostatic interactions in water, the release is as faster as the release observed with paclitaxel. These observations show that it is possible to develop a new drug-eluting stent capable of simultaneous delivering of an anti-proliferative agent and an anti-platelet drug. However, further research is required on this topic. | ||
| Fig. 6 Representative standard HPLC chromatograms of: (a) dipyridamole-and (b) paclitaxel. | ||
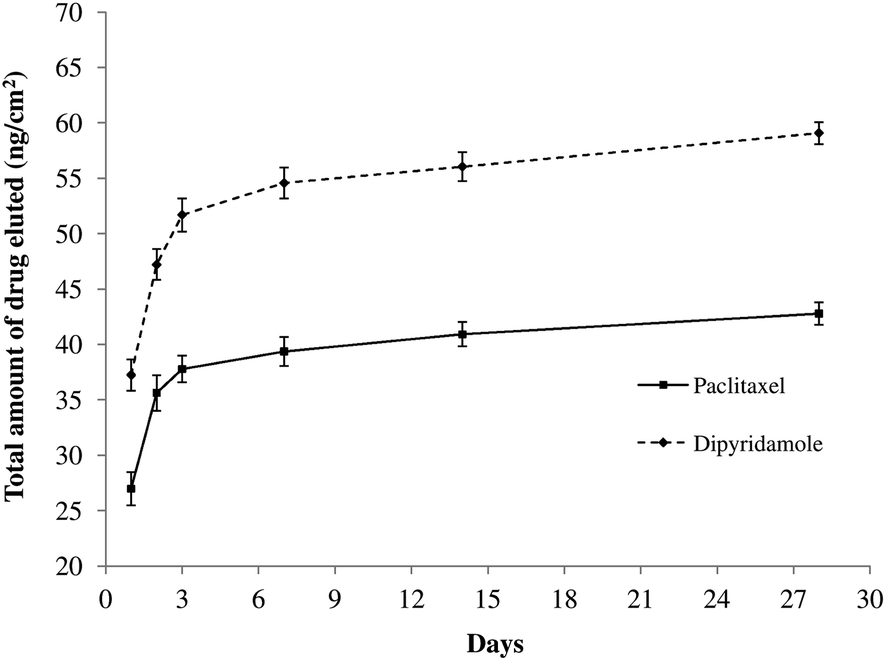 | ||
| Fig. 7 Dipyridamole and paclitaxel in vitro release profiles. | ||
Conclusions
The purpose of the current study was to study simultaneous delivery of dipyridamole (as a platelet inhibitor) and paclitaxel (as an anti-proliferative agent) from gold surfaces coated with 12-mercaptododecanoic acid and 1-nonanethiol mixed self-assembled monolayers. Samples were characterized by SEM, XPS, AFM, FTIR and CA measurements. AFM imaging showed that the mixed monolayer was well ordered and compact. XPS confirmed successful attachment of therapeutic agents onto the mixed self-assembled monolayer. The peak at 404.7 ± 0.2 eV for the sample containing dipyridamole (C24H40N8*O4) and paclitaxel (C47H51N*O14) was assigned to nitrogen atoms in the molecular structure of drugs. Results obtained from the SEM analysis revealed that the morphology of the monolayer coated by dipyridamole and paclitaxel was uniform. FTIR and CA techniques provided additional evidence for the successful attachment of drugs to the mixed monolayer. To demonstrate that the resulting structures can be used as drug delivery carriers, drug release in PBS was studied using HPLC. Interestingly, the release profiles of two drugs were similar in shape. These findings are promising for development of an advanced generation of DES. However, further studies are recommended.Acknowledgements
The authors would like to acknowledge The University of Tabriz for the financial support of this work.Notes and references
- M. A. Hanson, M. T. Fareed, S. L. Argenio, A. O. Agunwamba and T. R. Hanson, Primary Care, 2013, 40, 1 CrossRef PubMed.
- S. Mendis, P. Puska and B. Norrving, Global atlas on cardiovascular disease prevention and control, World Health Organization, Geneva, 2011 Search PubMed.
- W. Khan, S. Farah and A. J. Domb, J. Controlled Release, 2012, 161, 703 CrossRef CAS PubMed.
- Y. Li, R. Bhindi and L. M. Khachigian, J. Mol. Med., 2011, 89, 545 CrossRef CAS PubMed.
- G. Mani, M. D. Feldman, D. Patel and C. Agrawal, Biomaterials, 2007, 28, 1689 CrossRef CAS PubMed.
- C. D. Moyer, P. B. Berger and C. J. White, in Cardiovascular Medicine, ed. J. T. Willerson, J. N. Cohn, H. J. J. Wellens and D. R. Holmes, Springer, London, 2007, ch. 45, pp. 1031–1050 Search PubMed.
- K. K. Jain, Application of Biotechnology in Cardiovascular Therapeytics, Humana Press, NewYork, 2011 Search PubMed.
- R. Komatsu, M. Ueda, T. Naruko, A. Kojima and A. E. Becker, Circulation, 1998, 98, 224 CrossRef CAS.
- G. Mani, D. M. Johnson, D. Marton, M. D. Feldman, D. Patel, A. A. Ayon and C. M. Agrawal, Biomaterials, 2008, 29, 4561 CrossRef CAS PubMed.
- S. Garg and P. W. Serruys, J. Am. Coll. Cardiol., 2010, 56, S1 CrossRef CAS PubMed.
- S. Lancaster, S. Kakade and G. Mani, Langmuir, 2012, 28, 11511 CrossRef CAS PubMed.
- J. P. Chen, D. Hou, L. Pendyala, J. A. Goudevenos and N. G. Kounis, JACC: Cardiovascular Interventions, 2009, 2, 583 CrossRef PubMed.
- M. Joner, A. V. Finn, A. Farb, E. K. Mont, F. D. Kolodgie, E. Ladich, R. Kutys, K. Skorija, H. K. Gold and R. Virmani, J. Am. Coll. Cardiol., 2006, 48, 193 CrossRef PubMed.
- J. R. Nebeker, R. Virmani, C. L. Bennett, J. M. Hoffman, M. H. Samore, J. Alvarez, C. J. Davidson, J. M. McKoy, D. W. Raisch, B. K. Whisenant, P. R. Yarnold, S. M. Belknap, D. P. West, J. P. Gage, R. E. Morse, G. Gligoric, L. Davidson and M. D. Feldman, J. Am. Coll. Cardiol., 2006, 47, 175 CrossRef PubMed.
- Y. Otsuka, N. A. Chronos, R. P. Apkarian and K. A. Robinson, J. Invasive Cardiol., 2007, 19, 71 Search PubMed.
- J. Iqbal, J. Gunn and P. W. Serruys, Br. Med. Bull., 2013, 106, 193 CrossRef CAS PubMed.
- F. Nazneen, G. Herzog, D. W. M. Arrigan, N. Caplice, P. Benvenuto, P. Galvin and M. Thompson, J. Biomed. Mater. Res., Part B, 2012, 100, 1989 CrossRef PubMed.
- H. C. Gwon, J. Y. Hahn, K. W. Park, Y. B. Song, I. H. Chae, D. S. Lim, K. R. Han, J. H. Choi, S. H. Choi, H. J. Kang, B. K. Koo, T. Ahn, J. H. Yoon, M. H. Jeong, T. J. Hong, W. Y. Chung, Y. J. Choi, S. H. Hur, H. M. Kwon, D. W. Jeon, B. O. Kim, S. H. Park, N. H. Lee, H. K. Jeon, Y. Jang and H. S. Kim, Circulation, 2012, 125, 505 CrossRef CAS PubMed.
- Y. S. Lee, Self-Assembly and Nanotechnology: A Force Balance Approach, Wiley-Interscience, New Jersy, 2008 Search PubMed.
- S. Patil, S. Patil, S. Gawali, S. Shende, S. Jadhav and S. Basu, RSC Adv., 2013, 3, 19760 RSC.
- K. Yan, P. Li, H. Zhu, Y. Zhou, J. Ding, J. Shen, Z. Li, Z. Xu and P. K. Chu, RSC Adv., 2013, 3, 10598 RSC.
- Y. Wang, X. Li, G. Wu, J. Chen, Y. Wang, H. Gao and J. Ma, RSC Adv., 2013, 3, 13859 RSC.
- J. Zeng, P. Du and P. Liu, RSC Adv., 2013, 3, 19492 RSC.
- G. Mani, N. Torres and S. Oh, Biointerphases, 2011, 6, 33 CrossRef CAS PubMed.
- A. Mahapatro, D. M. Johnson, D. N. Patel, M. D. Feldman, A. A. Ayon and C. M. Agrawal, Langmuir, 2006, 22, 901 CrossRef CAS PubMed.
- A. Mirmohseni, J. Hosseini, M. Shojaei and S. Davaran, Colloids Surf., B, 2013, 112, 330 CrossRef CAS PubMed.
- J. Leonardi-Bee, P. M. Bath, M. G. Bousser, A. Davalos, H. C. Diener, B. Guiraud-Chaumeil, J. Sivenius, F. Yatsu and M. E. Dewey, Stroke, 2005, 36, 162 CrossRef CAS PubMed.
- C. D. d'Esterre and T. Y. Lee, Ann. N. Y. Acad. Sci., 2010, 1207, 71 CrossRef CAS PubMed.
- E. K. Rowinsky and R. C. Donehower, N. Engl. J. Med., 1995, 332, 1004 CrossRef CAS PubMed.
- W L. Hunter, US Pat., 0 074 934, 2010.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS PubMed.
- E. Ostuni, L. Yan and G. M. Whitesides, Colloids Surf., B, 1999, 15, 3 CrossRef CAS.
- C. D. Bain, E. B. Troughton, Y. T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321 CrossRef CAS.
- W. C. Tsai and P. J. R. Pai, Microchim. Acta, 2009, 166, 115 CrossRef CAS.
- G. Xu, D. P. Woodruff, N. Bennet, M. Elliott and J. E. Macdonald, Langmuir, 2010, 26, 8174 CrossRef CAS PubMed.
- R. Schweiss, D. Pleul, F. Simon, A. Janke, P. B. Welzel, B. Voit, W. Knool and C. Werner, J. Phys. Chem. B, 2004, 108, 2910 CrossRef CAS.
- N. K. Chaki, M. Aslam, J. Sharma and K. Vijayamohanan, J. Chem. Sci., 2001, 113, 659 CrossRef CAS.
- C. Kaufmann, G. Mani, D. Marton, D. Johnson and C. Agrawal, J. Biomed. Mater. Res., Part B, 2011, 98, 280 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |