
Improved synthesis of tadalafil using dimethyl carbonate and ionic liquids†
Abstract
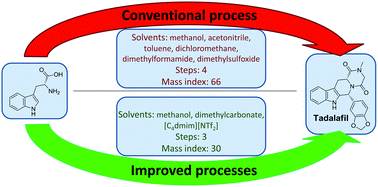
An improved synthesis of tadalafil, a drug for the treatment of male erectile dysfunction, involves the use of safer solvents and reagents as well as a reduced number of steps.
 Please wait while we load your content...
Please wait while we load your content...

