 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrosion behaviour of mild steel in 1-alkyl-3-methylimidazolium tricyanomethanide ionic liquids for CO2 capture applications†
Igor S. Molchan*a,
George E. Thompsonb,
Robert Lindsaya,
Peter Skeldona,
Vlassis Likodimos*b,
George Em. Romanosb,
Polycarpos Falarasb,
Gabriela Adamovac,
Boyan Ilievc and
Thomas J. S. Schubertc
aCorrosion and Protection Centre, School of Materials, The University of Manchester, Manchester, M13 9PL, UK. E-mail: igor.molchan@manchester.ac.uk
bDivision of Physical Chemistry, Institute of Advanced Materials, Physicochemical Processes, Nanotechnology and Microsystems (IAMPPNM), NCSR “Demokritos”, 153 10 Aghia Paraskevi Attikis, Athens, Greece. E-mail: likodimo@chem.demokritos.gr
cIOLITEC, Ionic Liquids Technologies GmbH, Salzstraße 184, D-74076 Heilbronn, Germany
First published on 9th December 2013
Abstract
The corrosion behaviour of mild steel (MS) was systematically investigated as a function of the alkyl chain length in the cation of 1-alkyl-3-methylimidazolium tricyanomethanide ([Cnmim]TCM, n = 2, 4, 6 and 8) ionic liquids (ILs) with respect to their potential application as a structural material and solvents for CO2 capture plants respectively. The surface of MS was examined by scanning electron microscopy, energy dispersive X-ray spectroscopy and micro-Raman mapping before and after immersion testing at temperatures of 70 and 80 °C for durations varying from 1 hour to 10 days. Corrosion initiates at the sites of MnS inclusions on the surface of MS, resulting in the formation of cavities due to the MnS dissolution, which may be surrounded by corrosion products containing magnetite (Fe3O4) and maghemite (γ-Fe2O3). The amount of the corrosion products generated around the inclusion sites decreased with the increase of the cation alkyl chain length, following the order [C2mim]TCM > [C4mim]TCM > [C6mim]TCM ≈ [C8mim]TCM. This was attributed to the corrosion inhibition effect of the ILs through adsorption on the metal surface and blocking active sites, with the inhibition efficiency increasing with the alkyl chain length. The underlying mechanism was associated with corrosion processes at active sites on the MS surface, such as sulphide inclusions, in the presence of residual water and oxygen in the IL. It was shown that increase of the water content in the ILs to about 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm resulted in faster dissolution of the MnS inclusions. Finally, it was demonstrated that removal of oxygen from the IL significantly reduced the corrosion rate.
000 ppm resulted in faster dissolution of the MnS inclusions. Finally, it was demonstrated that removal of oxygen from the IL significantly reduced the corrosion rate.
Introduction
Ionic liquids have become a subject of extensive study in the last decade due to their unique properties and potential for diverse technological applications, including utilisation for the capture of CO2 generated during fossil fuel combustion in industrial processes.1 Amongst the enormous number of possible IL structures interacting physically with CO2, the ideal IL solvent should exhibit a plethora of beneficial physicochemical and thermodynamic properties, including high thermal stability and CO2 capture capacity, low viscosity and fast binary CO2/IL diffusion, together with high solubility and chemical stability in water. These properties must further be supplemented by the ease of synthesis/purification and competitive cost. Applied in a typical CO2 scrubbing process, ILs may offer distinct advantages compared with conventional amine-based solvents, in particular (i) elimination of evaporative losses, (ii) sustainability in the presence of acidic gases and oxygen and (iii) minimisation of energy dissipation required for regeneration, which may have tremendous environmental and economical impacts in the CO2 capture process. On the other hand, the practical deployment of physically absorbing ILs is severely impaired by their reduced CO2 capture capacity and rate, both arising from the inherently different mechanisms underlying CO2 capture compared with amine solvents, e.g. chemisorption compared with physisorption. In this respect, ILs may compete with amine solvents only if the size of the scrubber increases or if scrubbing occurs at elevated pressure. Both these parameters may significantly increase the capital cost of installation. Thus, the selection of materials for construction of the CO2 capture plant to minimise the cost and to ensure integrity of the plant facilities is of great importance.Current and potential industrial applications of ILs require understanding of their interactions with metallic materials. Corrosion is particularly important where ILs are in contact with metallic components such as pipes, valves, vessels and reaction chambers. However, despite numerous publications dedicated to the physicochemical properties and application aspects of ILs,13 corrosion of metals and alloys in ILs is not widely covered, while detailed investigations of the underlying corrosion mechanisms remain the subject of discussion.
Since metallic materials are considered as the main construction materials for IL-operated installations, the corrosion behaviour of metals and alloys in ILs attracts particular attention. Corrosion of steels, nickel alloys, brass, aluminium and magnesium alloys in a number of ILs is an area of growing interest. The corrosion of the alloys in the ILs depends on the nature of the cation and anion of the IL and testing conditions, e.g. temperature, applied potential and impurities. Table 1 lists selected studies on the corrosivity of ILs toward widely used alloys. Corrosion is a severe issue for conventional monoethanol amine (MEA) operated CO2 capture plants, due to the corrosive nature of CO2.14 Corrosion testing at a pilot plant under MEA operation revealed that stainless steel generally exhibited a good performance with the corrosion rate not exceeding several μm per year in certain locations of the plant.15 On the other hand, the corrosion rate of MS varied widely from 1 μm per year to over several mm per year depending on the location. MS typically contains 0.05 to 0.25% carbon and, due to relatively low cost, it is used where large amounts of steel are required, for example, in construction, cars, fences and domestic goods. Hence, the use of MS is preferred for building CO2 capture IL-operated installations to reduce material costs if corrosion processes are effectively alleviated. Further, an increasing number of publications report on the inhibiting properties of ILs in aggressive environments. The addition of relatively small amounts of selected ILs to hydrochloric or sulphuric acid solutions, typically of the order of tens of mmol l−1 or hundred ppm, results in an increased inhibition efficiency of mild steel against corrosion to a value above 90% due to chemisorption or physisorption of the ILs on the alloy surface and the blocking of active sites.16,17
| Subject | Materials | Ionic liquids | Ref. |
|---|---|---|---|
| Formation of protective coating by immersion in ILs | Pure magnesium, AZ31 magnesium alloy | Tri(hexyl)tetradecylphosphoniumbis(trifluoromethanesulfonyl)amide; trihexyl(tetradecyl)phosphonium-bis-2,4,4-trimethylpentylphosphinate | 2 |
| Fundamental and applied study of corrosion of magnesium alloys in ILs | AZ91D magnesium alloy | [C4mim]trifluoromethylsulfonate; [C4mim]bis (trifluoromethylsulfonyl) | 3 |
| Study of ILs corrosivity in flow systems | Carbon steel, stainless steel (eqn (304)), nickel-based alloy, CuZn40 brass, copper, AlMg3 aluminium alloy | [C2mim]ethylsulphate; [C4mim]octylsulphate; C4mim]Cl; 1,3-dimethyl-imidazolium dimethylphosphate; (C12–C18) alkylpoly-(3)oxyethyldihydroxy-ethylmethyl-ammonium methylsulphate); cocosaklylpentaethoxi methyl ammonium methylsulphate | 4 |
| Study of room-temperature corrosion in ILs | Nickel, copper, 316 stainless steel | Mixture of AlCl3 and [C2mim]Cl | 5 |
| Application of soft X-ray SEM, combined with micro-spot X-ray absorption spectroscopy and X-ray fluorescence spectroscopy for in-situ investigation of corrosion in ILs | Nickel | 1-Butyl-1-methyl-pyrrolidiniumbis(trifluoromethylsulfonyl)amide | 6 |
| Study of the relationship between corrosion rate and structure of the ILs | 1018 carbon steel | ([C4mim]Cl; [C6mim]PF6; [C8mim]PF6; [C4mim] bis-(trifluoromethanesulfonyl)imide | 7 |
| Corrosion study of ILs with potential lubricant properties | CuSn8P bronze, 100Cr6 steel | Cholinebis(trifluoromethyl-sulfonyl)imide; choline methanesulfonate; butyl-trimethyl-ammonium bis(trifluoromethyl-sulfonyl)imide | 8 |
| Corrosion study at elevated temperature (70–325 °C) | Copper, nickel, 1018 carbon steel, brass, Inconel 600 | [C4mim]bis-(trifluoromethanesulfonyl)imide | 9 |
| Corrosion study at high temperature (220 °C) for applications in thermal circuits | 304 stainless steel and 1018 carbon steel | [C6mim]tri(pentafluoroethyl)trifluorophosphate; 1-butyl-1-methylpyrrolidinium trifluoromethanesulfonate; ethyl-dimethyl-propyl-ammonium bis(trifluoromethylsulphonyl)imide; 1-butyl-1-methylpyrrolidinium bis (trifluoromethylsulphonyl)imide | 10 |
| Corrosion study of potential lubricant ILs for aluminium alloys | AA6061, AA7075 and AA2011 aluminium alloys | [Cnmim]X (X = BF4, n = 2, 6 and 8. X = CF3SO3, n = 2. X = (4-CH3C6H4SO3), n = 2. X = PF6, n = 6. 1-butyl-3-methylpyridinium bis(trifluoromethylsulfonyl)imide | 11 |
| Corrosion behaviour in alkanolamine/IL emulsions for CO2 capture | 1020 carbon steel | [C4mim]BF4, [C2mim]BF4, [C2mim] trifluoromethanesulfonate | 12 |
ILs with tricyanomethanide (TCM) anion possess several beneficial properties, including low viscosity, low melting point, high thermal stability and electrical conductivity, with the ability of scale-up production.18 The ILs with the TCM anion are not widely investigated and have been mainly explored for the development of non-volatile, “solvent-free” electrolytes for dye-sensitised solar cells.19,20 Very recently, evaluation of the CO2 capture capacity of [Cnmim]TCM ILs in the form of liquid solvents as well as upon imbibition in ceramic porous supports has shown that TCM-based ILs, apart from their intrinsically low viscosity, afford significant CO2 solubility and selectivity which is higher than that of fluorinated ILs such as [Cnmim]PF6 (Fig. 1a and b).21 Most importantly, recent studies carried out by the authors have concluded that the CO2 absorption capacity and diffusivity can be markedly enhanced in binary [Cnmim]TCM/H2O systems in contrast to the commonly detrimental influence of water in most physically and chemically CO2 absorbing ILs (Fig. 1c and d).22
 | ||
| Fig. 1 (a and b) Comparative CO2 absorption capacity between [C4mim]TCM and [C4mim]PF6 at (a) low pressures and (b) high pressures at different temperatures. (c and d) Enhancement effect of water on the CO2 absorption capacity of the (c) [C8mim]TCM and (d) [C4mim]TCM; the water molar fraction is shown in the legends. | ||
In this work, the corrosion properties of [Cnmim]TCM ILs were investigated as a function of the alkyl chain length (n = 2, 4, 6 and 8), using mild steel as reference substrate, a low-cost building material in CO2 capture facilities. Systematic immersion tests of MS have been performed at elevated temperatures, 70 and 80 °C, for durations from 1 h to 10 days and increased water contents up to 5 wt%. The surface characteristics of MS before and after corrosion tests were evaluated in detail by scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDX) and micro-Raman mapping allowing new insights to the origin of the corrosion processes on the surface of MS in the TCM-based ILs.
Experimental
Mild steel (0.13–0.18% C, max. 0.40% Si, 0.7–0.9% Mn, max. 0.05% S, max. 0.05% P and Fe balance) was supplied by RS Components (UK) as a rod of 10 mm diameter. The rod was cut into disks of approximately 4 mm thickness, which were mechanically polished normal to the rolling direction using, consecutively, P240, P600, P1200 and P4000 wet emery papers followed by 3 and 1 μm diamond pastes on a velvet pad with a polyethylene glycol-based lubricant. The resultant roughness, Ra, measured by a Contour GT (Veeco) white light interferometer, was approximately 8 nm.The four types of ILs of purity over 98 mol% used for testing, included the following: (i) 1-methyl-3-ethylimidazolium tricyanomethanide, [C2mim]TCM, (ii) 1-methyl-3-butylimidazolium tricyanomethanide, [C4mim]TCM, (iii) 1-methyl-3-hexylimidazolium tricyanomethanide, [C6mim]TCM, and (iv) 1-methyl-3-octylimidazolium tricyanomethanide, [C8mim]TCM. The 1-alkyl-3-methylimidazolium tricyanomethanide ionic liquids were synthesised according to the experimental procedures established at Iolitec. 1 mol of the corresponding imidazolium-based ionic liquid chloride, [Cnmim]Cl (n = 2, 4, 6, 8) of purity over 98 mol% (Iolitec), was dissolved in 1.5 l of dry dichloromethane. 1.05 mol of sodium tricyanomethanide was added immediately to the dissolved chloride and the reaction mixture was stirred for 48 h at room temperature. The mixture was then filtered through celite; the mother liquor evaporated under reduced pressure and the product dried under high vacuum for 24 h at 40 °C. Ion chromatography showed that the chloride content of all products was below 0.5 mol%. The experimental yield of the reaction, i.e. the percentage of the theoretical yield, in all cases exceeded 90%. The water contents in the ILs measured using a C20 Coulometric Karl Fischer Titrator (Mettler Toledo) were 1050, 980, 265 and 265 ppm for n = 2, 4, 6 and 8 in [Cnmim]TCM respectively.
The MS specimens were sealed individually with each IL in autoclavable plastic bottles in the ambient atmosphere. Additionally, in order to confirm the influence of oxygen on IL's corrosivity, the specimen of MS was sealed with [C4mim]TCM in a nitrogen atmosphere. Prior to sealing, the IL was purged with nitrogen for 3 hours to remove air, the origin of oxygen. 4 ml of IL per specimen was used in the individual studies. The immersion testing was carried out at 70 and 80 °C for durations ranging from 1 h to 10 days, and at room temperature for 1 day. The temperatures up to 80 °C were selected to ensure long-term stability of the ILs. It was revealed that ILs change colour after keeping at 100 °C for 1 day that may be attributed to chemical transformations in the ILs; in contrast, no significant colour change was observed at 80 °C. In order to study the effect of water content in ILs on the IL corrosivity, [C2mim]TCM and [C8mim]TCM with increased water contents were prepared by adding eight droplets of water, with the approximate weight of an individual droplet being 0.025 g, to 4 ml of each IL using a pipette, followed by mixing at room temperature. The resultant water contents, determined by Karl Fischer titration, were 48900 and 53900 ppm for [C2mim]TCM and [C8mim]TCM respectively. The ILs used in the present study are miscible with water to a certain extent,22 and mixing led to the formation of a uniform liquid in several minutes.
A Zeiss Ultra-55 scanning electron microscope was employed for examination of the surface and local chemistry of the specimens. The microscope is equipped with SE1, BSE and EDX detectors. Secondary electron images were recorded at accelerating voltage varied from 2 to 15 kV. The acceleration voltage was adjusted to 8 kV for EDX analysis.
Micro-Raman spectra were recorded in backscattering configuration on a Renishaw inVia Reflex microscope using an Ar+ ion laser (λ = 514.5 nm, 2.41 eV) and a high-power near infrared diode laser (λ = 785 nm, 1.58 eV) as excitation sources. The laser beam was focused onto the specimens by means of a 100× (NA = 0.85) objective at a laser power below 0.3 mW in order to avoid local heating and phase transformation of the iron oxides.23 Analysis of the scattered beam was performed on a 250 mm focal length spectrometer along with a 1800 lines per mm diffraction grating and a high-sensitivity CCD detector. Raman mapping was implemented on a motorized feedback-controlled XYZ mapping specimen stage that allowed area mapping with a step size of 0.1 μm in the point-by-point mode.
Results and discussion
Immersion testing in as-received ILs
The specimens of MS were immersed in the four [Cnmim]TCM ILs at 80 °C for 1, 3, 5 and 10 days. Fig. 2 shows representative scanning electron micrographs of the MS specimens after immersion for 1 day. After immersion in [C2mim]TCM, the MS surface reveals numerous craters (Fig. 2a). | ||
| Fig. 2 Scanning electron micrographs of the surface of MS after immersion in (a) [C2mim]TCM, (b) [C4mim]TCM, (c) [C6mim]TCM and (d) [C8mim]TCM at 80 °C for 1 day. | ||
Individual craters consist of a cavity of circular or elongated shape and size up to ∼1 μm that is surrounded by a darker region of dimensions up to 3 μm. Craters of similar shapes are generated during immersion in [C4mim]TCM (Fig. 2b). The number of craters per unit area is similar to that in [C2mim]TCM and is of the order of 1 × 106 cm−2; however, the crater dimensions are slightly reduced, with the maximum crater dimensions generally not exceeding 2 μm. Likewise immersion in [C2mim]TCM and [C4mim]TCM, craters are generated on the surface of MS after immersion in [C6mim]TCM (Fig. 2c). However, the crater dimensions are significantly smaller, generally up to 1 μm, with most of the craters having dimensions of several hundred nanometres. The appearance of the craters generated in [C8mim]TCM is similar to those generated in [C6mim]TCM (Fig. 2d). Most of the craters are of submicron dimensions; however, the size of individual craters may reach 2 μm. The estimated numbers of craters per unit area generated in the four ILs are approximately the same. The surface appearance of the MS does not change significantly with increase of immersion duration to 10 days (Fig. S1 of ESI†). Craters of similar shapes and numbers are evident after increased immersion duration. It may be summarised that the average dimensions of the craters generated in the ILs increases in the order [C8mim]TCM ≈ [C6mim]TCM < [C4mim]TCM < [C2mim]TCM. Interestingly, the order also follows the increase of the length of alkyl chain of the cation.
It is well known for both mild steel and stainless steel that pits are initiated at sulphide inclusions, e.g. MnS.24,25 Sulphur impurities are present in all steels and due to the low solubility of sulphides, sulphide inclusions are formed. In carbon steels, the attack initiates in the matrix close to the sulphide inclusion, which is more noble than the matrix. Indeed, local EDX analysis of the surface of the as-polished MS shown in Fig. 3a confirmed the presence of manganese and sulphur in the inclusion sites of darker appearance (Fig. 3b). The local EDX spectrum recorded outside of the metal matrix area (Fig. 3c) exhibited only peaks of iron and carbon corresponding to the alloy constituents (hydrocarbon contamination on the surface may also contribute to the carbon yield). Detailed EDX analysis within the typical crater area of MS immersed in [C2mim]TCM for 10 days (Fig. 4a) is displayed in Fig. 4b and c. The oxygen peak centred at 0.52 keV suggests the presence of corrosion products, e.g. iron oxides or hydroxides, in the crater; no oxygen was detected outside the crater, indicating that corrosion products are confined within the crater area. A weak silicon peak in the EDX spectra originates from the alloy constituents. Silicon is typically added to steel during the production process for the purpose of deoxidisation and may be in the form of SiO2; thus, SiO2 may also contribute to the oxygen peaks in the spectra. It was shown elsewhere that sulphide inclusions may exist as separate particles or shells surrounding an oxide core.26 Sulphides undergo dissolution during corrosion, whereas oxides remain intact. Manganese and sulphur were not detected in the craters, suggesting that the MnS inclusion was completely dissolved. EDX analysis of individual craters generated in [C6mim]TCM and [C8mim]TCM after 10 days of immersion did not reveal the presence of manganese and sulphur. No oxygen was detected in most of the craters, suggesting that the rate of corrosion product formation in [C6mim]TCM and [C8mim]TCM is significantly reduced compared with [C2mim]TCM. SEM/EDX examination of MS after immersion in ILs for 1, 3 and 5 days revealed similar crater shapes and numbers per unit area compared with those generated after 10 days of immersion. Manganese and sulphur were not detected in the analysed craters after immersion in [C2mim]TCM, [C4mim]TCM and [C6mim]TCM. Thus, most MnS inclusions are dissolved within 1 day of immersion. A weak manganese peak was detected in one of the craters generated in [C8mim]TCM after one day of immersion, possibly due to incomplete dissolution of the inclusion.
 | ||
| Fig. 3 (a) Scanning electron micrograph of as-polished mild steel and local EDX spectra recorded in the (b) point A and (c) point B of the micrograph. | ||
 | ||
| Fig. 4 Scanning electron micrograph and related local EDX spectra of MS after immersion in [C2mim]TCM at 80 °C for 10 days: (a) micrograph; (b) spectrum recorded in the cavity in the location marked A; (c) spectrum recorded in the area surrounding the cavity marked B. | ||
Since the surface of MS was affected to a greater extent during immersion in [C2mim]TCM compared with the three other ILs, [C2mim]TCM was selected to carry out an immersion test at 80 °C for a shorter immersion period to reveal the time necessary for complete dissolution of the MnS inclusions at this temperature. The durations of the immersion tests varied from 1 to 8 h; Fig. 5 displays the surface appearance of MS after 1 and 3 h of immersion. Corrosion products were identified around the MnS inclusions immersed for 1 h, revealing oxygen peaks in the related EDX spectra (Fig. 5a). MnS inclusions are located in the centres of the craters. The narrow gaps separating inclusions and the metallic matrix suggest that the inclusions started to dissolve. After 3 h, most of the inclusions are partly dissolved. The gap between the inclusion remnants and metallic matrix is significantly wider than after 1 h; complete dissolution may take place in the small craters, leaving cavities of dimensions of several hundred nanometres (Fig. 5b). Further increase of the immersion duration revealed that the inclusions are totally dissolved in [C2mim]TCM within approximately 8 h at 80 °C. It is well established that pit initiation may start at a certain temperature that is reproducible.27 Immersion of MS in [C2mim]TCM at room temperature for 1 day did not cause any significant changes on the surface; the surface appearance was similar to that of the as-polished alloy (Fig. S2 of ESI†). Thus, temperature plays an important role in increasing the rates of dissolution of MnS inclusions and formation of corrosion products in the selected ILs.
 | ||
| Fig. 5 Scanning electron micrographs of the surface of MS after immersion in [C2mim]TCM at 80 °C for (a) 1 h and (b) 3 h. | ||
The chemical nature, phase composition and spatial distribution of the corrosion products on the surfaces of MS after immersion in the [Cnmim]TCM ILs at 80 °C for 1 and 10 days were subsequently investigated by micro-Raman spectroscopy combined with local area mapping. Fig. 6 shows representative Raman spectra at different points on the surface of MS after immersion in [C2mim]TCM for 1 and 10 days, using the 514.5 nm laser line. An intense Raman signal was systematically detected in the numerous craters developed on the polished MS surface after immersion in [C2mim]TCM for both 1 and 10 days that is consistent with the SEM/EDX analysis. Extended Raman spectra acquired locally on the crater areas revealed the presence of three asymmetric broad bands around 350, 535 and 672 cm−1 together with two higher frequency broad modes at ∼1400 and ∼1600 cm−1 at excitation wavelength of 514.5 nm as shown in Fig. 7a. These bands can be mainly attributed to the presence of the iron-deficient, inverse spinel phase of maghemite (γ-Fe2O3)28,29 that was further supported by direct comparison with the Raman spectra of representative reference iron oxides (Fig. 7a), where the contribution of the parent magnetite spinel (Fe3O4) to the observed Raman spectra is also evidenced. Specifically, the superposition of the most intense A1g mode of magnetite at 669 cm−1,30 with the broad maghemite band at 720 cm−1, may account for the observed asymmetric line shape that peaks at 672 cm−1 with a shoulder at higher frequencies, while similar effects can be applied to rationalise the broad line shapes and deviations of the observed peak frequencies from the corresponding ones of single phase maghemite or magnetite.29,31 This indicates that the phase composition of the corrosion products on the MS surface can be essentially attributed to a mixture of maghemite and magnetite phases, although quantification of their relative amounts is rather difficult due to the diverse electronic structures of the two constituent iron oxides (semiconducting γ-Fe2O3 compared with semimetallic Fe3O4 at room temperature) and the concomitant variation of their Raman scattering cross section under 514.5 nm laser excitation.32 In fact, changing the excitation wavelength from 514.5 to 785 nm, further from the maghemite absorption edge at about 600 nm,33 resulted in a Raman spectrum dominated by the bands of magnetite (Fig. 7b), whose optical response varies only weakly in the corresponding wavelength range.34 This behaviour is characteristic of the (near-) resonance Raman effect as the excitation energy at 514.5 nm (2.41 eV) approaches the bandgap of maghemite (∼2.0 eV), causing the enhancement of the Raman modes of γ-Fe2O3 over those of Fe3O4 that, however, prevail under non-resonant excitation conditions at 785 nm.
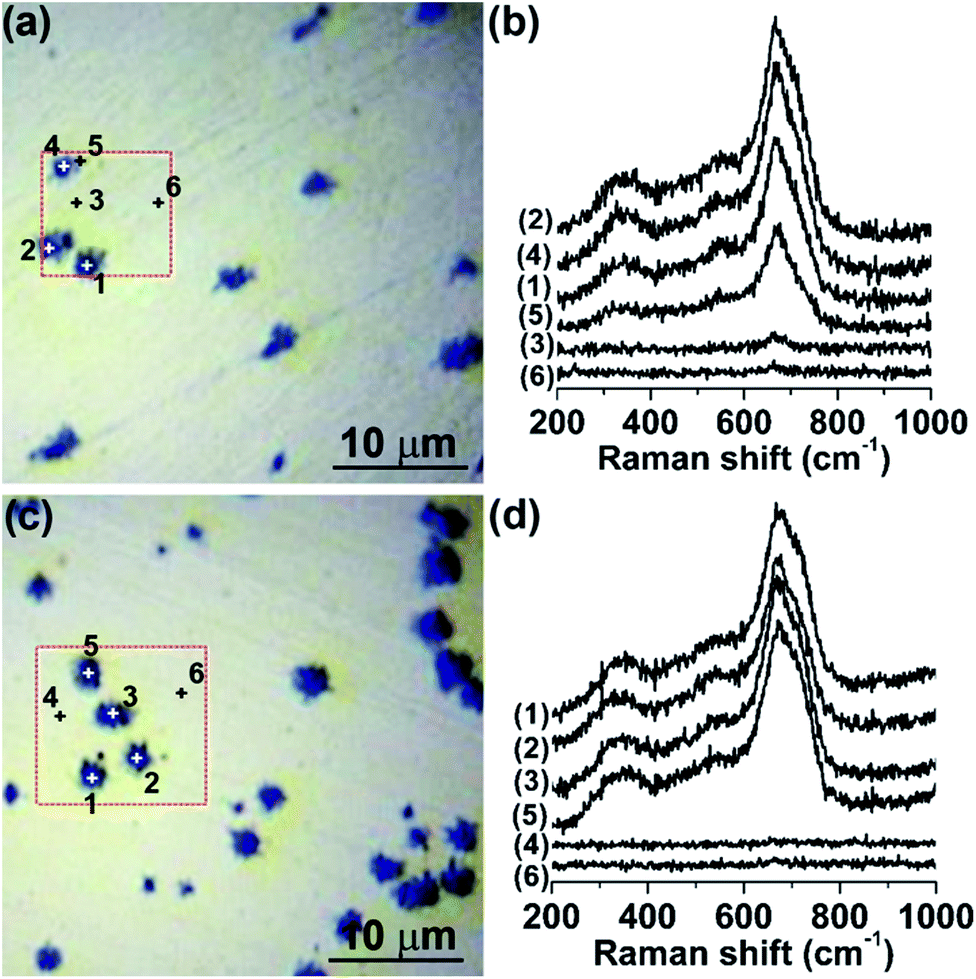 | ||
| Fig. 6 (a and c) Optical micrographs and (b and d) related micro-Raman spectra at different points on the surface of MS after immersion in [C2mim]TCM for (a and b) 1 and (c and d) 10 days at 80 °C; excitation wavelength is 514.5 nm. | ||
 | ||
| Fig. 7 (a) Comparison of the Raman spectra of corrosion products on MS after immersion in [C2mim]TCM at 80 °C for 10 days with reference samples of maghemite (γ-Fe2O3), magnetite (Fe3O4), hematite (α-Fe2O3) and goethite (α-FeOOH); excitation wavelength is 514.5 nm. (b) Comparison of the Raman spectra of corrosion products on MS and magnetite recorded at excitation wavelength of 785 nm. | ||
Raman mapping of the integrated area of the most intense mode at 672 cm−1 at excitation wavelength of 514.5 nm has been subsequently employed to probe the spatial distribution of the corrosion products on the MS surface after immersion in [C2mim]TCM for 10 days (Fig. 8). Comparison of the Raman maps with the corresponding optical images revealed that the maximum Raman intensity occurs around the craters as well as at regions in-between neighbouring craters, reflecting the uneven iron oxide distribution around the craters in perfect agreement with the EDX spatial analysis. Raman spectra were accordingly acquired at different points on the MS surface after immersion in [C4mim]TCM for 1 and 10 days, at 514.5 nm (Fig. S3 of ESI†). Likewise for [C2mim]TCM, the composite Raman spectra of mixed γ-Fe2O3/Fe3O4 spinels were identified at regions in the dark areas surrounding the MS craters, whose densities were similar to those upon [C2mim]TCM immersion.
 | ||
| Fig. 8 Raman mapping of (a–d) single and (e–h) neighbouring craters on the MS surface after immersion in [C2mim]TCM at 80 °C for 10 days: (a and e) optical images, (b and f) Raman maps of the integrated area of the 672 cm−1 band, (c and g) superpositions of the optical and Raman images and (d and h) Raman spectra corresponding to the maximum (red) and minimum (black) signal on the Raman maps. Excitation wavelength is 514.5 nm. | ||
However, spatial Raman analysis of the MS specimens after immersion in [C6mim]TCM and [C8mim]TCM, revealed a significant reduction in the extent of corrosion product formation (Fig. 9). The Raman spectra of the γ-Fe2O3/Fe3O4 corrosion products were progressively weaker on the considerably smaller dark regions surrounding cavities on the MS surface for [C6mim]TCM and [C8mim]TCM compared with those obtained for [C2mim]TCM and [C4mim]TCM. Moreover, for many craters, the Raman signal was markedly reduced or totally suppressed, indicating that the growth of γ-Fe2O3/Fe3O4 corrosion layers was considerably impeded in the presence of [C6mim]TCM and [C8mim]TCM that is in agreement with the EDX analysis. An intensity of the iron oxides Raman peak associated with the individual crater marked (6) in Fig. 9(b) is relatively high; however, the intensities of the peaks of the most of the craters are significantly reduced compared with those of [C2mim]TCM. Comparative Raman maps of MS craters after immersion in [C4mim]TCM and [C8mim]TCM for 10 days (Fig. 10) verified directly the marked decrease of the γ-Fe2O3/Fe3O4 Raman intensity, although the spatial distributions of the iron oxides were similar for both cases. This behaviour confirmed that corrosion product formation on MS is considerably hindered for the less dense and more viscous [C6mim]TCM and [C8mim]TCM ILs with the longer alkyl chain length of the cation.
 | ||
| Fig. 9 Optical images and Raman spectra at different points on the surface of MS after immersion in (a and b) [C6mim]TCM and (c and d) [C8mim]TCM at 80 °C for 10 days; excitation wavelength is 514.5 nm. | ||
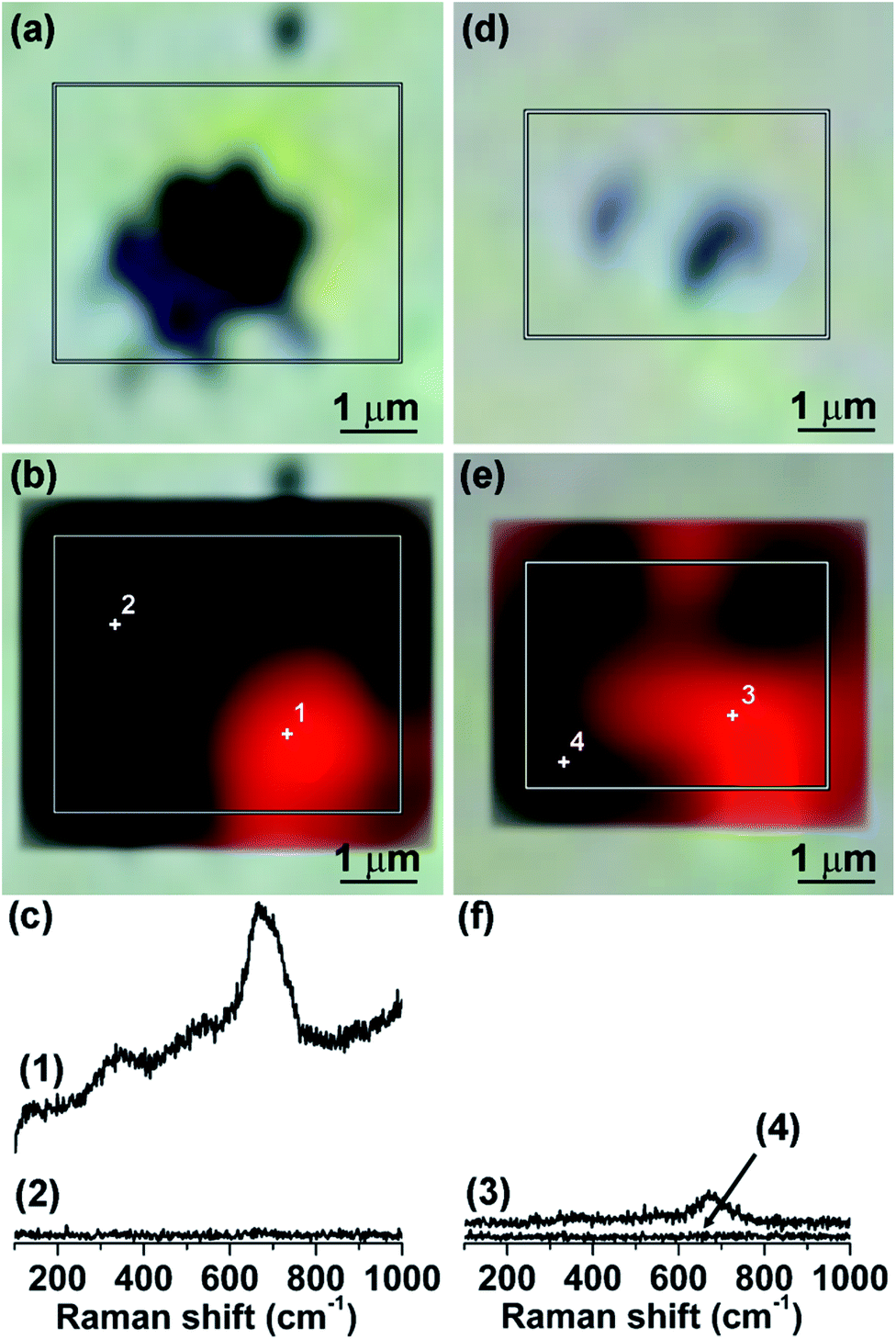 | ||
| Fig. 10 Raman mapping of the craters on the MS surface after immersion in (a–c) [C4mim]TCM and (d–f) [C8mim]TCM at 80 °C for 10 days, at excitation wavelength of 514.5 nm: (a and d) optical image, (b and e) Raman map of the integrated area of the 672 cm−1 band and (c and f) Raman spectra corresponding to the maximum (red) and minimum (black) signal on the Raman maps at the same intensity scale. | ||
Based on the SEM/EDX and micro-Raman analyses, it is inferred that the main effect on the MS surface after immersion in the [Cnmim]TCM ILs for 1 h to 10 days at 80 °C is the dissolution of sulphide inclusions (MnS), leading to cavity formation without substantial dissolution of the surrounding metal matrix. The process is accompanied by the generation of γ-Fe2O3/Fe3O4 corrosion products in the cavities and the surrounding areas of the MnS inclusions that ceases as soon as the inclusions are dissolved. The increase of the craters size as well as the formation of corrosion products were particularly pronounced for [C2mim]TCM and [C4mim]TCM. On the other hand, both the crater dimensions and the amount of corrosion products were significantly reduced for [C6mim]TCM and [C8mim]TCM, unveiling a marked dependence of the [Cnmim]TCM ILs corrosive ability on the cation alkyl chain length, which largely determines their hydrophobicity; the increase of the alkyl chain length increases hydrophobicity of the ILs.35 In fact, the water contents measured by Karl Fischer titration decreased with increase of the length of the cation alkyl chain, suggesting that the progressive effectiveness of the [Cnmim]TCM ILs in the dissolution of sulphide inclusions and the formation of corrosion products in MS may be associated with the presence of small amounts of water in ILs and possibly dissolved oxygen.
Immersion tests in ILs with increased water content
In order to study the role of water in the dissolution of MnS inclusions and the formation of corrosion products, immersion tests of MS in the ILs with increased water contents were carried out at 70 °C for two hours. The ILs with the highest and the lowest corrosivities, [C2mim]TCM and [C8mim]TCM respectively, with water contents of approximately 50000 ppm were selected. Fig. 11 compares the surface appearance of MS after immersion in [C2mim]TCM and [C8mim]TCM with and without added water. Craters with cavities surrounded by corrosion products of darker appearance are evident on the surface of MS immersed in [C2mim]TCM (Fig. 11a and b). The majority of the craters generated in the as-received [C2mim]TCM are filled with inclusions. EDX analysis of several inclusions identified them as MnS. Immersion in [C2mim]TCM with added water resulted in complete dissolution of MnS inclusions in most craters. Craters were not evident widely on the surface of MS after immersion in [C8mim]TCM (Fig. 11c); the MS surface appearance was similar to the as-received alloy. Darker features on the surface were identified as MnS by local EDX analyses. Similar to [C2mim]TCM with 48900 ppm water, immersion in [C8mim]TCM with 53900 ppm water led to the complete dissolution of MnS inclusions in the majority of craters (Fig. 11d). Local EDX analysis of several craters did not reveal the presence of oxygen, thus, confirming that the amount of corrosion products generated in [C8mim]TCM is insignificant. Weak sulphur peaks were detected in a few craters suggesting that MnS inclusion remnants remain in individual craters.
 | ||
| Fig. 11 Scanning electron micrographs of the surface of MS after immersion at 70 °C for 2 h in: (a) as-received [C2mim]TCM, (b) [C2mim]TCM with 48900 ppm water, (c) as-received [C8mim]TCM and (d) [C8mim]TCM with 53900 ppm water. | ||
Fig. 12 shows representative Raman spectra on the MS surface after immersion in [C2mim]TCM with and without added water at 70 °C for 2 h. In the case of the as-received [C2mim]TCM, the formation of mixed γ-Fe2O3/Fe3O4 corrosion products was directly identified by the corresponding Raman spectra at the dark areas surrounding the MS craters. However, a series of additional broad Raman bands at 217, 277, 317, 475 and 590 cm−1 were systematically detected at the regions of brighter appearance in the craters centres, superimposed with the iron oxide Raman spectra (Fig. 12a). These modes can be primarily attributed to the inherently weak Raman spectra of the cubic MnS phase (alabandite), especially the broad band at 590 cm−1.36 The relatively intense bands at 277, and 317 cm−1 may indicate the contribution of wurtzite type Raman modes, similar to buseckite (Fe, Zn, Mn)S sulphides, possibly due to the partial dissolution/transformation of the MnS inclusions.37 On the other hand, no evidence of sulphide inclusions was traced by Raman mapping on the abundant craters covering the MS surface after immersion in [C2mim]TCM with 48900 ppm water (Fig. 12b), corroborating the total dissolution of MnS in the presence of water. Furthermore, Raman analysis of MS after immersion in as-received [C8mim]TCM at 70 °C for 2 h revealed the presence of the MnS inclusions in the few craters generated on MS, together with the absence of any iron oxide products (Fig. S4a and b of ESI†). Moreover, no sulphide inclusions or corrosion products were identified on the MS craters after immersion in [C8mim]TCM with 53900 ppm water by micro-Raman (only an enhanced background signal) (Fig. S4c and d of ESI†), providing further evidence for the total dissolution of MnS upon increasing the ILs water content.
 | ||
| Fig. 12 (a and c) Optical images and (b and d) Raman spectra at different points on the surface of MS after immersion in (a and b) as-received [C2mim]TCM and (c and d) [C2mim]TCM with 48900 ppm water at 70 °C for 2 h; excitation wavelength is 514.5 nm. | ||
Corrosion mechanism
The molecules of the ILs used in the present study do not contain oxygen atoms. Thus, the only way to form oxides in the areas adjacent to the inclusions is through their interaction with water–oxygen dissolved in the ILs during manufacture and storage, with additional intake during immersion testing. Visual observation of the IL droplet placed in the SEM chamber through a built-in video camera during pumping down revealed intense gas evolution from the droplet, providing a clear indication for the presence of dissolved air–water in the IL. Therefore, it was assumed that corrosion events in the ILs proceed similarly to those in water-containing media. The study of corrosion of steels in moist media reviewed previously by Wranglen24,25 was used for the explanation of MS corrosion in the ILs. The sulphide inclusions were classified as active or non-active with regard to pit initiation. It was proposed that the active MnS inclusions are surrounded by a metallic matrix with submicroscopic MnS precipitates, often referred to as a sulphur-infected matrix that was generated during the steel production process. The sulphur-infected matrix near the MnS inclusions is highly active due to the large contact area between the fine precipitates and the iron; such regions form anodic sites. MnS inclusions, fine MnS precipitates surrounding inclusions and the non-infected metal matrix act as cathodes. Fine precipitates have significantly higher solubility compared with the larger inclusions. Dissolution of the precipitates is accompanied by the increase of acidity with the formation of H2S and HS− ions that catalyse both anodic dissolution of iron and hydrogen evolution at the cathodic sites. The large-area metallic matrix, not contaminated by sulphides, serves as a cathode, providing oxygen reduction. MnS inclusions and surrounding precipitates also act as cathodic sites, however, mainly for hydrogen evolution. Development of the acidity due to hydrolysis of iron ions released at anodic areas facilitates dissolution of the inclusions.Pure water is not corrosive to most metals. However, dissolved gases, e.g., oxygen, carbon dioxide, sulfur dioxide and impurities, e.g. chloride ions, can cause corrosion attack. In many cases, pits initiate at physical or chemical inhomogenities on the surface, such as inclusions, flaws, second-phase intermetallic particles, grain boundaries, etc., and temperature may be a critical factor.38 No corrosion was identified after 1 day of immersion in [C2mim]TCM at room temperature, suggesting that the corrosion rate at room temperature is sufficiently low. Considering that ILs contain up to 2 mol% impurities, including up to 0.5 mol% chlorides, dissolved water and oxygen that may cause corrosion events, it can be assumed that IL adsorbed on the metal surface may act as a barrier to the initiation of corrosion at room temperature. At elevated temperature, probably after reaching a certain value, iron starts oxidising in the anodic areas, i.e. in the sulphur-infected matrix, thus generating corrosion products. Indeed, after 1 h of immersion in [C2mim]TCM at 80 °C, corrosion products are evident at the areas surrounding MnS inclusions. Since MnS is easily dissolved in acidic media,25 inclusions start to dissolve chemically due to the increase of the local acidity as a result of the hydrolysis of metal ions. Addition of approximately 50000 ppm water to [C2mim]TCM and [C8mim]TCM facilitates dissolution of the inclusions; complete dissolution of the majority of the inclusions occurs within 2 h of immersion at 70 °C. The IL probably assists in prevention of cavity development, i.e. growth, propagation and coalescence of the neighbouring cavities, by adsorption on the surface through chemisorption or physisorption, depending on the value of the standard free energy of adsorption. The adsorption is stronger for the IL with increased length of the alkyl chain in the cation. The increase of the inhibition effect with increase of the size of IL molecule was reported elsewhere.16 The effect was attributed to the greater surface coverage by the larger molecule during the chemisorption process. The rates of iron dissolution and corrosion product formation in [C6mim]TCM and [C8mim]TCM are significantly reduced at the sites of MnS inclusions and adjacent sulphur-infected matrix leading to reduced coverage with corrosion products compared with those generated in [C2mim]TCM and [C4mim]TCM.
Perissi et al.10 estimated the amount of the dissolved oxygen in the ILs for solar collector applications, studying corrosion of AISI 1018 and AISI 304 steels in several ILs at elevated temperatures. It was determined that the fraction of oxygen in the IL may be three times larger than in water, which can affect corrosion processes. In order to confirm that oxygen may be responsible for corrosion initiation in the ILs with TCM anions, the two MS specimens were individually sealed with the relatively corrosive [C4mim]TCM IL in air and in nitrogen; in the latter case, the IL was purged with nitrogen to ensure that most dissolved air is removed. The sealed specimens were then maintained at 80 °C for 1 h. The micrographs of Fig. 13 show the typical appearance of the craters generated under air and nitrogen atmosphere. The MnS inclusion generated in air-sealed conditions is surrounded by corrosion products of darker appearance that was confirmed by local EDX analysis (Fig. 13a). In contrast, no corrosion products are evident around the MnS inclusion on the surface of MS after immersion in nitrogen atmosphere (Fig. 13b).
 | ||
| Fig. 13 Scanning electron micrographs of the surface of MS after immersion in [C2mim]TCM at 80 °C for 1 h: (a) sealed in air; (b) sealed in nitrogen atmosphere after purging IL with nitrogen for 3 h. | ||
Thus, the main chemical and electrochemical reactions involved in corrosion of MS in [Cnmim]TCM ILs may be as follows. The anodic process proceeds through dissolution of iron according to the reaction:
| Fe → Fe2+ + 2e−. | (1) |
The main cathodic process occurs at the matrix regions that are not contaminated by sulphur through reduction of oxygen dissolved in the IL by electrons produced by the anodic reaction:
| O2 + 2H2O + 4e− → 4OH−. | (2) |
This process is accompanied by a further cathodic reaction on MnS inclusions and the sulphur-infected matrix resulting in localised generation of hydrogen:
| 2H+ + 2e− → H2. | (3) |
Dissolution of sulphides proceeds through the reaction:
| MnS + 2H+ → H2S + Mn2+. | (4) |
FeOH2+ ions, formed after subsequent hydrolysis and oxidation of the Fe2+ ions, are finally precipitated as magnetite, detected by Raman mapping:
| 2FeOH2+ + Fe2+ + 2H2O → Fe3O4 + 6H+. | (5) |
Further, Fe3O4 is partly oxidised to γ-Fe2O3:
| 3Fe3O4 + 0.75O2 → 4.5γ-Fe2O3. | (6) |
This may proceed through oxidation of the outer part of the Fe3O4 layer, thus generating a two-layered oxide with a Fe3O4 inner layer and a γ-Fe2O3 outer layer.39
Finally, it may be concluded that in the [Cnmim]TCM ILs, corrosion products composed of magnetite and maghemite are generated due to the interaction of iron with oxygen dissolved in the ILs at the anodic sites in the presence of residual water. On the other hand, the [Cnmim]TCM ILs are inferred to inhibit corrosion in MS by adsorption on the metal surface,40 with their inhibition efficiency increasing markedly with the increase of the of alkyl chain length in the cation and the concomitant increase of hydrophobicity. Further, adsorption of the IL molecules at active sites in MS prevents propagation of the corrosion processes in the bulk of the alloy once the inclusion is dissolved, supporting the potential use of MS for the construction of low cost CO2 capture plant installations.
Conclusions
The corrosivity of 1-alkyl-3-methylimidazolium tricyanomethanide ILs, as promising solvents for CO2 capture, was investigated as a function of the cation alkyl chain length using mild steel, a relatively low cost metallic material, by immersion tests at elevated temperatures. Detailed surface analysis by SEM/EDX examination verified that the surface morphology of mild steel changes in all [Cnmim]TCM (n = 2, 4, 6 and 8) ILs at a temperature of 80 °C during immersion from 1 to 10 days. The morphological changes initiated in the areas adjacent to MnS inclusions and were accompanied by the generation of craters comprising cavities due to the dissolution of MnS inclusions, surrounded by corrosion products of variable extents, depending on the specific IL. The phase composition of the corrosion products was identified by micro-Raman mapping to be mixed magnetite–maghemite oxides forming preferentially at the areas surrounding the inclusion sites. Corrosion products in [C2mim]TCM and [C4mim]TCM were generated within the first hour of immersion at 80 °C, whereas no significant changes to the surface morphology was observed after immersion in [C8mim]TCM for 2 h at 70 °C. The finding suggests that cations with longer alkyl chains reduce significantly the dissolution rate of MnS inclusions. The crater densities and shapes did not changed markedly with the increase of immersion time up to 10 days, confirming that corrosion processes become significantly slower after the MnS precipitates and inclusions are dissolved.The coverage of the mild steel surface with corrosion products defined as the craters dimensions increased in the order [C8mim]TCM ≈ [C6mim]TCM < [C4mim]TCM < [C2mim]TCM. Further, the magnetite–maghemite Raman intensity was greater for the denser and less viscous [C2mim]TCM and [C4mim]TCM ILs compared with [C6mim]TCM and [C8mim]TCM ILs that were significantly less corrosive. Increase of the water contents up to approximately 50000 ppm in [C2mim]TCM and [C8mim]TCM resulted in faster dissolution of the MnS inclusions.
Corrosion in MS by the [Cnmim]TCM ILs was concluded to be driven by two processes, specifically (i) corrosion initiation at the MnS inclusion sites due to the interaction with dissolved oxygen in the presence of residual water, accompanied by dissolution of MnS and (ii) inhibition of corrosion by adsorption of the IL molecules at the active sites on the metal surface. The corrosion inhibition efficiency was found to be significantly enhanced for the [Cnmim]TCM ILs with the longer length of alkyl chain in the cation (n = 6, 8). Increase of the alkyl chain length in the cation in combination with oxygen-free environment may significantly reduce corrosion attack and propagation in MS, thus ensuring surface integrity and the preservation of the materials mechanical properties. This is specifically important for the selection of low cost structural materials for CO2 plant installations.
Acknowledgements
The research was supported by EC in the framework of the IOLICAP Grant (Project no. 283077; Call Identifier FP7-ENERGY-2011-1).References
- (a) N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shahb and P. Fennell, Energy Environ. Sci., 2010, 3, 1645 RSC; (b) M. Hasib-ur-Rahmana, M. Siaj and F. Larachi, Chem. Eng. Process., 2010, 49, 313 CrossRef PubMed; (c) J. F. Brennecke and B. E. Gurkan, J. Phys. Chem. Lett., 2010, 1, 3459 CrossRef CAS.
- (a) N. Birbilis, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Surf. Coat. Technol., 2007, 201, 4496 CrossRef CAS PubMed; (b) P. C. Howlett, T. Khoo, G. Mooketsi, J. Efthimiadis, D. R. MacFarlaneb and M. Forsyth, Electrochim. Acta, 2010, 55, 2377 CrossRef CAS PubMed.
- (a) A. Shkurankov, S. Z. E. Abedin and F. Endres, Aust. J. Chem., 2007, 60, 35 CrossRef CAS; (b) S. Caporali, F. Ghezzi, A. Giorgetti, A. Lavacchi, A. Tolstogouzov and U. Bardi, Adv. Eng. Mater., 2007, 9, 185 CrossRef CAS.
- M. Uerdingen, C. Treber, M. Balser, G. Schmitt and C. Werner, Green Chem., 2005, 7, 321 RSC.
- P.-C. Lin, I.-W. Sun, J.-K. Chang, C.-J. Su and J.-C. Lin, Corros. Sci., 2011, 53, 4318 CrossRef CAS PubMed.
- B. Bozzini, A. Gianoncelli, B. Kaulich, M. Kiskinova, C. Melea and M. Prasciolu, Phys. Chem. Chem. Phys., 2011, 13, 7968 RSC.
- M. F. Arenas and R. G. Reddy, J. Min. Metall., Sect. B, 2003, 39, 81 CrossRef CAS.
- (a) C. Gabler, C. Tomastik, J. Brenner, L. Pisarova, N. Doerr and G. Allmaier, Green Chem., 2011, 13, 2869 RSC; (b) L. Pisarova, C. Gabler, N. Dorr, E. Pittenauer and G. Allmaier, Tribol. Int., 2012, 46, 73 CrossRef CAS PubMed.
- (a) I. Perissi, U. Bardi, S. Caporali and A. Lavacchi, Corros. Sci., 2006, 48, 2349 CrossRef CAS PubMed; (b) A. B. Tolstoguzov, U. Bardi and S. P. Chenakin, Bull. Russ. Acad. Sci.: Phys., 2008, 72, 605 CrossRef.
- I. Perissi, U. Bardi, S. Caporali, A. Fossatia and A. Lavacchi, Sol. Energy Mater. Sol. Cells, 2008, 92, 510 CrossRef CAS PubMed.
- M.-D. Bermudez, A.-E. Jimenez and G. Martinez-Nicolas, Appl. Surf. Sci., 2007, 253, 7295 CrossRef CAS PubMed.
- M. Hasib-ur-Rahman, H. Bouteldja, P. Fongarland, M. Siaj and F. Larachi, Ind. Eng. Chem. Res., 2012, 51, 8711 CrossRef CAS.
- (a) S. Aparicio, M. Atilhan and F. Karadas, Ind. Eng. Chem. Res., 2010, 49, 9580 CrossRef CAS; (b) M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS PubMed; (c) Ionic Liquids – New Aspects for the Future, ed. J.-i. Kadokawa, InTech, Croatia, 2013, p. 695 Search PubMed.
- (a) K. S. George and S. Nesic, Corrosion, 2007, 63, 178 CrossRef CAS; (b) S. Nesic, M. Nordsveen, R. Nyborg and A. Stangeland, Corrosion'2001, NACE International, Houston, TX, 2001, p. 01040 Search PubMed.
- J. Kittel, R. Idem, D. Gelowitz, P. Tontiwachwuthikul, G. Parrain and A. Bonneau, Energy Procedia, 2009, 1, 791 CrossRef CAS PubMed.
- N. V. Likhanova, M. A. Dominguez-Aguilar, O. Olivares-Xometl, N. Nava-Entzana, E. Arce and H. Dorantes, Corros. Sci., 2010, 52, 2088 CrossRef CAS PubMed.
- (a) T. Tuken, F. Demir, N. Kicir, G. Sıgircik and M. Erbil, Corros. Sci., 2012, 59, 110 CrossRef CAS PubMed; (b) A. Zarrouk, M. Messali, M. R. Aouad, M. Assouag, H. Zarrok, R. Salghi, B. Hammouti and A. Chetouani, J. Chem. Pharm. Res., 2012, 4, 3427 CAS; (c) H. Ashassi-Sorkhabi and M. Es'haghi, Mater. Chem. Phys., 2009, 114, 267 CrossRef CAS PubMed.
- (a) S. A. Forsyth, S. R. Batten, Q. Dai and D. R. MacFarlane, Aust. J. Chem., 2004, 57, 121 CrossRef CAS; (b) H. Brand, J. F. Liebman, A. Schulz, P. Mayer and A. Villinger, Eur. J. Inorg. Chem., 2006, 4294 CrossRef CAS; (c) Y. Yoshida, K. Muroi, A. Otsuka, G. Saito, M. Takahashi and T. Yoko, Inorg. Chem., 2004, 43, 1458 CrossRef CAS PubMed; (d) T. J. Wooster, K. M. Johanson, K. J. Fraser, D. R. MacFarlane and J. L. Scott, Green Chem., 2006, 8, 691 RSC.
- M. Bidikoudi, T. Stergiopoulos, V. Likodimos, G. E. Romanos, M. Francisco, B. Iliev, G. Adamová, T. J. S. Schubert and P. Falaras, J. Mater. Chem. A, 2013, 1, 10474 CAS.
- M. Marszalek, Z. Fei, D.-R. Zhu, R. Scopelliti, P. J. Dyson, S. M. Zakeeruddin and M. Grätzel, Inorg. Chem., 2011, 50, 11561 CrossRef CAS PubMed.
- (a) A. I. Labropoulos, G. E. Romanos, E. Kouvelos, P. Falaras, V. Likodimos, M. Francisco, M. C. Kroon, B. Iliev, G. Adamova and T. J. S. Schubert, J. Phys. Chem. C, 2013, 117, 10114 CrossRef CAS; (b) O. C. Vangeli, G. E. Romanos, K. G. Beltsios, D. Fokas, E. P. Kouvelos, K. L. Stefanopoulos and N. K. Kanellopoulos, J. Phys. Chem. B, 2010, 114, 6480 CrossRef CAS PubMed; (c) O. Tzialla, C. Veziri, X. Papatryfon, K. G. Beltsios, A. Labropoulos, B. Iliev, G. Adamova, T. J. S. Schubert, M. C. Kroon, M. Francisco, L. F. Zubeir, G. E. Romanos and G. N. Karanikolos, J. Phys. Chem. C, 2013, 117, 18434 CrossRef CAS.
- G. E. Romanos, L. F. Zubeir, V. Likodimos, P. Falaras, M. C. Kroon, B. Iliev, G. Adamova and T. J. S. Schubert, J. Phys. Chem. B, 2013, 117, 12234 CrossRef CAS PubMed.
- K. S. K. Varadwaj, M. K. Panigrahi and J. Ghose, J. Solid State Chem., 2004, 177, 4286 CrossRef CAS PubMed.
- G. Wranglen, Corros. Sci., 1974, 14, 331 CrossRef.
- G. Wranglen, Corros. Sci., 1969, 9, 585 CrossRef CAS.
- M. Smialowski, Z. Szklarska-Smialowska, M. Rychcik and A. Szummer, Corros. Sci., 1969, 9, 123 CrossRef CAS.
- (a) R. Ovarfort, Corros. Sci., 1988, 28, 139 CrossRef; (b) R. Ovarfort, Corros. Sci., 1989, 29, 987 CrossRef.
- (a) N. Boucherit, A. Hugot-Le-Goff and S. Joiret, Corros. Sci., 1991, 32, 497 CrossRef CAS; (b) D. L. A. de Faria, S. Venancio Silva and M. T. de Oliveira, J. Raman Spectrosc., 1997, 28, 873 CrossRef CAS; (c) Ph. Colomban, S. Cherifi and G. Despert, J. Raman Spectrosc., 2008, 39, 881 CrossRef CAS.
- I. Chourpa, L. Douziech-Eyrolles, L. Ngaboni-Okassa, J.-F. Fouquenet, S. Cohen-Jonathan, M. Souce, H. Marchais and P. Dubois, Analyst, 2005, 10, 1395 RSC.
- (a) S. Joiret, M. Keddam, X. R. Nóvoa, M. C. Pérez, C. Rangel and H. Takenouti, Cem. Concr. Compos., 2002, 24, 7 CrossRef CAS; (b) O. N. Shebanova and P. Lazor, J. Solid State Chem., 2003, 174, 424 CrossRef CAS; (c) C. Gabrielli, S. Joiret, M. Keddam, H. Perrot, N. Portail, P. Rousseau and V. Vivier, J. Electrochem. Soc., 2006, 153, B68 CrossRef CAS PubMed.
- Z. Cvejic, S. Rakic, A. Kremenovic, B. Antic, C. Jovalekic and P. Colomban, Solid State Sci., 2006, 8, 908 CrossRef CAS PubMed.
- A. I. Kontos, V. Likodimos, T. Stergiopoulos, D. S. Tsoukleris, P. Falaras, I. Rabias, G. Papavassiliou, D. Kim, J. Kunze and P. Schmuki, Chem. Mater., 2009, 21, 662 CrossRef CAS.
- N. J. Cherepy, D. B. Liston, J. A. Lovejoy, H. Deng and J. Z. Zhang, J. Phys. Chem. B, 1998, 102, 770 CrossRef CAS.
- N. Guskos, G. J. Papadopoulos, V. Likodimos, S. Patapis, D. Yarmis, A. Przepiera, K. Przepiera, J. Majszscyk, J. Typek, M. Wabia, K. Aidinis and Z. Drazek, Mater. Res. Bull., 2002, 37, 1051 CrossRef CAS.
- M. G. Freire, C. M. S. S. Neves, P. J. Carvalho, R. L. Gardas, A. M. Fernandes, I. M. Marrucho, L. M. N. B. F. Santos and J. A. P. Coutinho, J. Phys. Chem. B, 2007, 111, 13082 CrossRef CAS PubMed.
- (a) C. Ma, J. R. Beckett and G. R. Rossman, Am. Mineral., 2012, 97, 2056 CrossRef CAS; (b) RRUFF Raman spectral database, http://rruff.info.
- C. Ma, J. R. Beckett and G. R. Rossman, Am. Mineral., 2012, 97, 1226 CrossRef CAS.
- G. S. Frankel, J. Electrochem. Soc., 1998, 145, 2186 CrossRef CAS PubMed.
- M. Nagayama and M. Cohen, J. Electrochem. Soc., 1962, 109, 781 CrossRef CAS PubMed.
- C. Y. Peñalber, G. A. Baker and S. Baldelli, J. Phys. Chem. B, 2013, 117, 5939 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra45872e |
| This journal is © The Royal Society of Chemistry 2014 |