Synthesis of TiO2 hollow spheres by selective etching of Au@TiO2 core–shell nanoparticles for dye sensitized solar cell applications†
Min-kyeong Song,
Prabhakar Rai,
Kyeong-Jun Ko,
Seung-Ho Jeon,
Bum-Soo Chon,
Chung-Hyun Lee and
Yeon-Tae Yu*
Division of Advanced Materials Engineering, Research Centre for Advanced Materials Development, College of Engineering, Chonbuk National University, Jeonju 561-756, South Korea. E-mail: yeontae@jbnu.ac.kr; Fax: +82-63-270-2305; Tel: +82-63-270-2288
First published on 28th November 2013
Abstract
Heterostructured Au@TiO2 core–shell nanoparticles (NPs) were synthesized by a microwave assisted hydrothermal method. A colloidal method was used to synthesize 40 ± 5 nm Au NPs, whereas a microwave-assisted hydrothermal method was used to deposit a TiO2 shell layer with 60 ± 10 nm shell thickness on Au NPs. The average size of TiO2 NPs was 17 ± 2 nm and size was increased with increasing reaction temperature without considerable change in shell thickness. The stability of Au@TiO2 core–shell NPs in iodide electrolyte solution was examined. It was found that the Au NPs are unstable in the iodide electrolyte and lost their surface plasmon resonance (SPR) characteristics. Hollow TiO2 NPs (150–200 nm in diameter) were produced by selective etching of as-prepared Au@TiO2 core–shell NPs in KCN solution. The final hollow TiO2 spheres were applied as a scattering layer on top of a nanocrystalline TiO2 film, serving as the photoanode of dye sensitized solar cells (DSCs). A high efficiency of 7.40% was achieved with TiO2 hollow spheres, compared with 5.21% for the electrode with commercial TiO2. It was also found that the efficiency increased with increasing crystallinity of TiO2 NPs. The increment in efficiency was related to efficient light scattering, electrolyte diffusing feasibility for better electron transport, and a high surface area for higher dye loading.
Introduction
DSCs were first demonstrated by O'Regan and Gratzel in 1991 and have been considered as a promising alternative to the conventional silicon-based solar cells because of their potentially low production cost and facile technology.1–13 Since then, they have been intensively investigated, however their relatively low efficiencies have not been improved significantly during the past two decades and the best efficiency is still below 12%. Much effort has been made toward improving the performance of DSCs by optimizing photosensitizers,2–5 photoanode,6–8 redox electrolyte,9–11 and counter electrode.12–14 It has been found that the structure and morphology of photoanode plays a critical role in determining the light to electricity conversion efficiency.15 Generally, a well performing photoanode requires high specific surface area, fast electron transport, and effective light scattering properties. Therefore, in most of DSCs, the photoelectrode is normally made of anatase TiO2 nanocrystals of ∼20 nm in diameter, to ensure a large internal surface area for loading large amounts of dye molecules. Meanwhile, the photoelectrode made of anatase TiO2 nanocrystals of ∼20 nm in diameter results in the transmittance of long wavelength (red) part of the incident light without exciting dye molecules.Therefore, intensive research on DSCs has been focused on achieving higher device efficiency and better performance by rationalizing the construction of the photoelectrodes to either speed up electron transfer or enhance light scattering.16,17 In recent years SPR of metal NPs has been regarded as an attractive approach to boost the performance of DSCs.18–23 For example, by coupling semiconductor nanostructures with metal NPs in a core–shell geometry one can observe an enhancement in their photo-conversion efficiency. The enhancement in photo-conversion efficiency is related to improved charge separation as a result of localized electromagnetic field, increased absorption due to surface plasmons, promoting electron transfer to adsorbed species, and electron storage effects. However, in most of the DSCs, fluid I−/I3− redox couples is used as an electrolyte which shows incompatibility with some metallic component materials.24 These I−/I3− redox couples can corrode the metals, such as Ag and Au, especially in the presence of water and oxygen. Therefore, use of metal NPs is not suitable for long term stability of DSCs device because in most cases shell materials are porous in nature and electrolyte can penetrate through shell and corrode the core material.25–28 Therefore, many researchers are focusing on enhance light scattering to enhance light harvesting.29–33 In most cases a second layer of either phosphor (like CeO2, Ce:Y3Al5O12) or TiO2 anatase particles of ∼400 nm in diameter is often used to scatter back the transmitted light, in order to enhance light harvesting.29–31 However, these large size particles only helps to enhance light scattering, with small contribution in light absorption because of low surface area for dye adsorbing. In other words, these large particles increase the total volume of the semiconductor photoanode, while contribute little in electron injection. Therefore, it is necessary to increase the light absorption by scattering effects and also maintain a high specific surface area of the photoanode films for efficient dye adsorption. Recently, nanoporous and/or mesoporous TiO2 submicrometer microspheres have been utilized as photoanodes in DSCs to achieve high energy conversion efficiencies.34–38
In this present work, we report the synthesis of TiO2 hollow spheres by using Au NPs as template. Au@TiO2 core–shell NPs were synthesized by microwave-assisted hydrothermal method and hollow TiO2 NPs were produced by selective etching Au NPs in KCN solution. The instability of Au NPs of Au@TiO2 core–shell NPs in iodide electrolyte showed that the use of surface plasmon enhanced solar cell is not useful for long term stability. Therefore, hollow TiO2 NPs were adopted as scattering layer and its effect on energy conversion efficiency of DSCs was investigated in detail.
Experimental
Material synthesis
A typical procedure for synthesizing the Au@TiO2 core–shell NPs was carried out as in the literature, but modified.25 1 mL of 0.01 M HAuCl4 solution (Sigma-Aldrich, USA) was mixed with 2 mL of 0.01 M sodium citrate solution (Yakuri, Japan). The mixture was stirred vigorously for 5 min. Then, 1.2 mL of 0.01 M ascorbic acid (Showa, Japan) was added dropwise. The mixture was turned orchid and then rapidly to reddish brown. After stirring continuously for 15 min, 3 mL of 0.04 M TiF4 solution (Aldrich, USA) was added. The mixture was subsequently diluted to 30 mL with deionized water and transferred to a microwave oven (CEM Mars 5) and heated for 5 minutes to reach the reaction temperature and maintained at that temperature (100–180 °C) with rapid stirring for 1–15 h. After completion of reaction, the products were cooled to room temperature and separated by centrifuge at 7000 rpm for 10 min, then washed with distilled water. The cycles of separation and washing were repeated 5 times to remove the remaining ions. The final samples were dried in an oven at 80 °C and collected for further use.The hollow TiO2 NPs were produced by selective etching of as-prepared Au@TiO2 core–shell NPs in KCN solution according to the reported literature.25,26 A 7.5 mL of 0.05 M KCN solution was added to as prepared Au@TiO2 core–shell NPs and stirred for 10 min. The pH value was modified to 10.5 using 0.01 M NaOH solution. The reaction mixture was stirred in air for 3 h. The resultant white suspension was separated by centrifuge at 9500 rpm for 15 min and then washed with distilled water. The cycles of separation and washing were repeated 5 times. The final white product was dried in an oven at 80 °C and then collected.
DSSC cell fabrication and efficiency measurements
TiO2 photoelectrode film of 6 μm thicknesses was created by squeeze printing of a commercial nanocrystalline TiO2 paste (ENB Korea Co.) on fluorine doped tin oxide (FTO) as shown in Fig. S1 (ESI†). Four different photoanode structures were studied: a monolayer and double layer of commercial nanocrystalline TiO2 film without any scattering layer and with a 2.5 μm hollow TiO2 sphere scattering layer. The pasted films were heat treated at 550 °C for 45 minutes for calcination and sintering. The films were dye loaded in a 0.3 mM N719 (B2, Dyesol) dye solution. Counter electrodes were prepared by Pt ion sputtering (E-1010, HITACHI) on FTO. Finally, 60 μm Surlyn (Dyesol) spacers were used to seal the cells. The cells were filled with an electrolyte composed of 0.6 M 1-butyl-3-methylimidazolium iodide (BMII), 0.1 M guanidiniumthiocyanate (GuSCN), 0.03 M I2, and 0.5 M 4-tertbutylpyridine (Aldrich) in cetonitrile–valeronitrile (85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15, vol%).
:15, vol%).
Material characterization
The transmission electron microscope (TEM; H-7650 HITACHI) images were taken using an accelerating voltage of 100 kV. The crystallographic structures of the solid samples were determined with D/Max 2005 Rigaku X-ray diffractometer equipped with graphite monochromatized high-intensity Cu Kα1 radiation (λ = 1.541 87 Å). The UV-visible, reflectance and transmittance spectra were conducted on a UV-visible spectroscope (UV-2550, Shimadzu). Surface area was analyzed by Brunauer–Emmett–Teller (BET) surface area analyzer (TriStar, Micromeritics). The cross section of photoanode was observed by field emission scanning electron microscopy (FESEM) (Hitachi S-4300). I–V curves of the DSCs was measured under AM1.5 G simulated solar light (100 mW cm−2) (Polaronix K3000).Results and discussion
Diffraction patterns of Au@TiO2 core–shell NPs synthesized at different temperatures are shown in Fig. 1a. It reveals that anatase phase of TiO2 is formed in all specimen. There is no phase transition observed in either TiO2 or Au crystals. However, crystallinity of TiO2 is increased according to synthesized temperatures. As can be seen from figure, the intensity of TiO2 (101) plane increases as compared to Au (111) plane. Furthermore, TiO2 crystallite size is calculated by Scherrer formula;| t = kλ/Bcosθ |
 | ||
| Fig. 1 Diffraction patterns of Au@TiO2 core–shell NPs prepared by microwave-assisted hydrothermal method; (a) for 1 h at different reaction temperatures (100–180 °C) and (b) at 180 °C for different reaction times (1–15 h). | ||
The morphology of the final products displayed in Fig. 2 represents the formation of core–shell NPs with Au NPs as a core while TiO2 as a shell. The Au NPs are well dispersed, spherical in shape and their size ranges from 40 ± 5 nm as shown in Fig. 2a. After hydrothermal reaction using TiF4 as precursor at 100 °C for 1 h, the formation of heterogeneous TiO2 layers on Au NPs occurs as exhibited in Fig. 2b. The over all size of Au@TiO2 core–shell NPs is about 200 ± 20 nm, where the TiO2 shell thickness is about 60 ± 10 nm. It can be clearly seen that the shell is composed of about 14 nm TiO2 NPs. Temperature-dependent morphological evolution at fixed time (1 h) is also collected to investigate formation of Au@TiO2 core–shell NPs, and the corresponding results obtained from an earlier reaction temperature (120–180 °C) are shown in Fig. 2c–f. It reveals that overall morphology of Au@TiO2 core–shell NPs is almost maintained except the particle size of primary TiO2 NPs, composing shell layer, increases with increasing synthesis temperatures. Similarly, time-dependent morphological evolution at fixed temperature (180 °C) is also carried out to investigate the formation of Au@TiO2 core–shell NPs (ESI, Fig. S3†). It reveals that particle size of primary TiO2 NPs increases with increasing synthesis time without considerable changes in morphology of Au@TiO2 core–shell NPs. As evident from TEM images, the well-defined TiO2 NPs are formed with increasing reaction time.
 | ||
| Fig. 2 TEM images of (a) Au NPs and Au@TiO2 core–shell NPs synthesized at (b) 100, (c) 120, (d) 140, (e) 160 and (f) 180 °C for 1 h. | ||
For DSCs application of as prepared Au@TiO2 core–shell NPs, it was necessary to examine the stability of Au NPs in iodide electrolyte. The stability of Au NPs in electrolyte solution is examined and shown in Fig. 3 (ESI, Fig. S4†). The SPR peaks of the Au NPs and Au@TiO2 core–shell NPs synthesized at 180 °C for 15 h is shown in Fig. 3a. The Au NPs absorption peak is recorded at 528 nm which is red-shifted to 585 nm after TiO2 shell formation. The red-shift in SPR peaks of the Au NPs in Au@TiO2 core–shell NPs is caused by the larger refractive index of TiO2 encapsulating the Au cores. When, Au@TiO2 core–shell NPs are added to electrolyte, immediately Au NPs dissolve and color of the solution changed from violet to white (ESI, Fig. S4†). The UV-visible absorption spectra also show that the characteristic SPR peak of Au NPs is disappeared. Electrolyte treated Au@TiO2 core–shell NPs are further examined by TEM and result is displayed in Fig. 3b. This TEM image clearly shows the formation of hollow TiO2 without Au NPs. This result reveals that TiO2 shell is porous in nature though which electrolyte reaches to Au NPs and causes its poisoning.
 | ||
| Fig. 3 (a) UV-visible spectra of Au and Au@TiO2 core–shell NPs before and after electrolyte treatment. (b) TEM image of Au@TiO2 core–shell NPs after electrolyte treatment. | ||
It has been reported by several researchers that halide ions, except F−, can form specific adsorption layer on the surface of Au NPs.39–43 Among various halides, the Au–I binding strength is highest and it decreases with decreasing atomic number; I > Br > Cl.39 However, the synthesis of Au NPs dispersed in aqueous media is generally carried out by HAuCl4 precursor, as in this study, and it is believed that the resulting NPs from this precursor would normally have adsorbed Cl− ions released from the precursor itself. This is possibly related to better alignment of Cl− ions with the underlying Au (111) planes causing little strain.40 However, the I− ion adsorption causes the greatest strain because of the largest mismatch with the Au (111) lattice plane. The I2 adsorption does create significant strain on the NPs surface leading to the deformation of the NPs and in some places aggregation.40 It is also reported that the addition of iodide ions probably leads to the fragmentation of Au NPs leading to the formation of small NPs.43 However, this study shows that dissolution of Au NPs also takes place and therefore hollow TiO2 NPs are formed. This dissolution of Au NPs in electrolyte solution is not well understood and need further investigation. Meanwhile, this study shows that specifically adsorbed iodide ions can inevitably change the surface states of Au NPs, which are critical for the enhancement of DSCs efficiency. Thus, the use of SPR of Au@TiO2 core–shell NPs in DSCs possibly increases its efficiency but it would not be useful for long term stability of DSCs. Therefore, we examined the effect of hollow TiO2 as light scattering layer on efficiency of DSCs.
The hollow TiO2 NPs were obtained from Au@TiO2 core–shell NPs by KCN etching. Diffraction pattern of hollow TiO2 NPs is exhibited in Fig. 4a. It reveals all the characteristic peaks of anatase TiO2, whereas no peaks related to Au NPs are recorded. It suggests that the Au NPs are dissolved by KCN and completely removed. It is also evident from Fig. 4b that Au NPs completely dissolved by KCN and 30–50 nm space (equal to Au NPs size) was left at the centre of the particles. This result further confirms that TiO2 shell is porous in nature and thus limits the application of SPR of Au@TiO2 core–shell NPs in DSCs because of its short term stability. Therefore, as prepared hollow TiO2 NPs are used for the fabrication of DSCs cell.
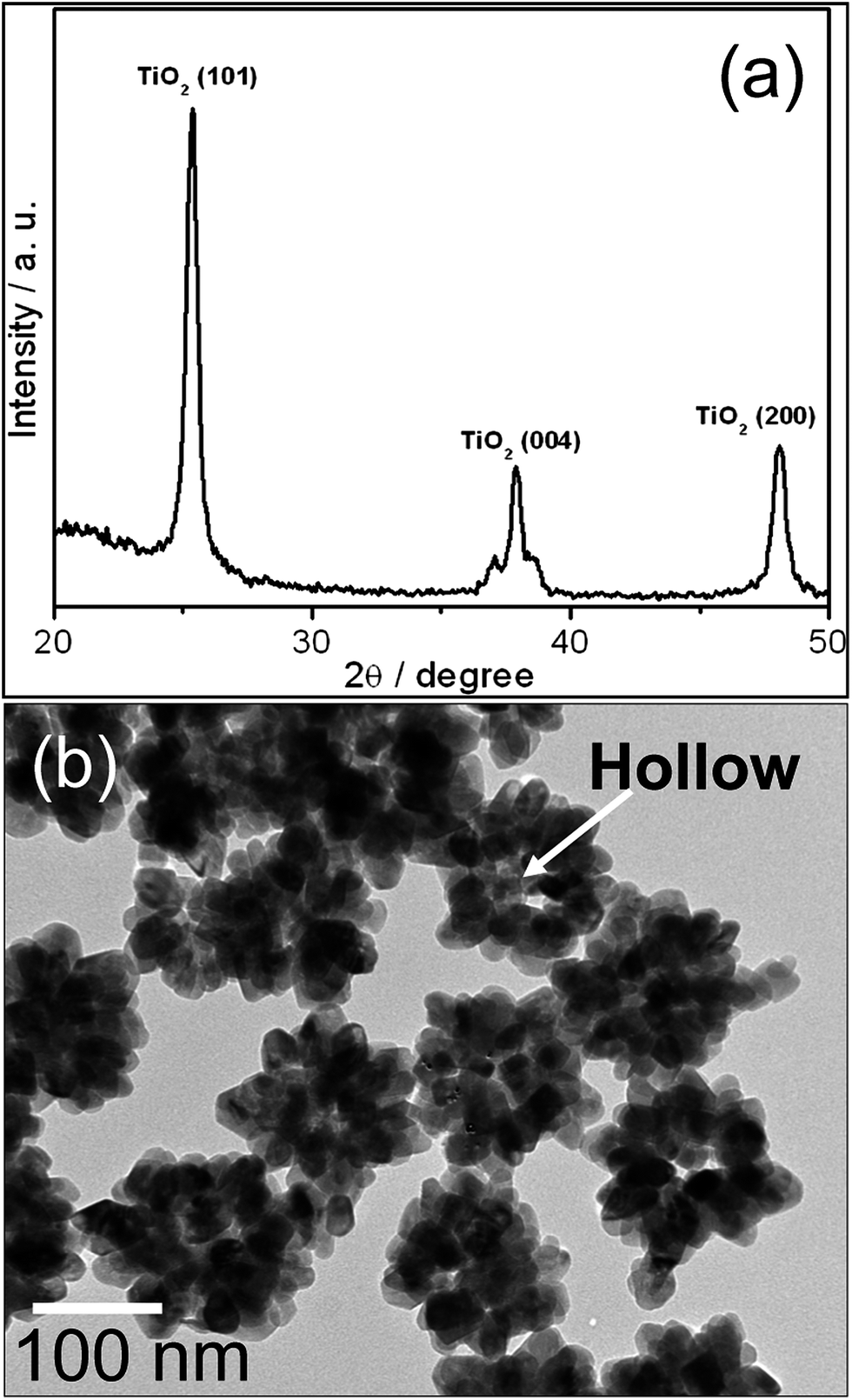 | ||
| Fig. 4 (a) XRD profile and (b) TEM image of hollow TiO2 NPs. | ||
To demonstrate the scattering effect of the hollow TiO2 spheres on the improvement of photovoltaic performance of DSCs, a bilayer photoelectrode was constructed by coating a layer of the hollow TiO2 NPs on top of commercial TiO2 as displayed in Fig. 5. As prepared devices were tested to compare with that of a commercial TiO2 photoelectrode-based device.
 | ||
| Fig. 5 Cross-sectional SEM image of TiO2 electrode (a) commercial TiO2 monolayer, (b) commercial TiO2 monolayer + hollow TiO2 monolayer, (c) commercial TiO2 double layer, (d) commercial TiO2 double layer + hollow TiO2 monolayer. | ||
The characteristic current (I)–voltage (V) curves are displayed in Fig. 6, showing that the short-circuit current (Jsc, 16.02 mA cm−2) for the DSCs with a bilayer photoelectrode is significantly improved in comparison with that for the pristine commercial TiO2 NPs based one (Jsc, 11.46 mA cm−2). As a result, the overall photo-conversion efficiency (η) of the hollow TiO2 DSCs achieves 7.40%, which is higher than the commercial TiO2 NPs based DSCs (5.21%) measured in parallel experiments by 1.42 fold. This short-circuit current is further improved for the DSCs with a tri-layer photoelectrode consisting of double layer of commercial TiO2 NPs and monolayer of hollow TiO2 (Jsc, 17.8 mA cm−2) in comparison with bilayer photoelectrode of commercial TiO2 NPs based one (Jsc, 13.7 mA cm−2). In this case, the photo-conversion efficiency (η) of the hollow TiO2 DSC achieves 7.63%, which is again higher than the commercial TiO2 NPs based DSC (6.66%) by 1.15 fold.
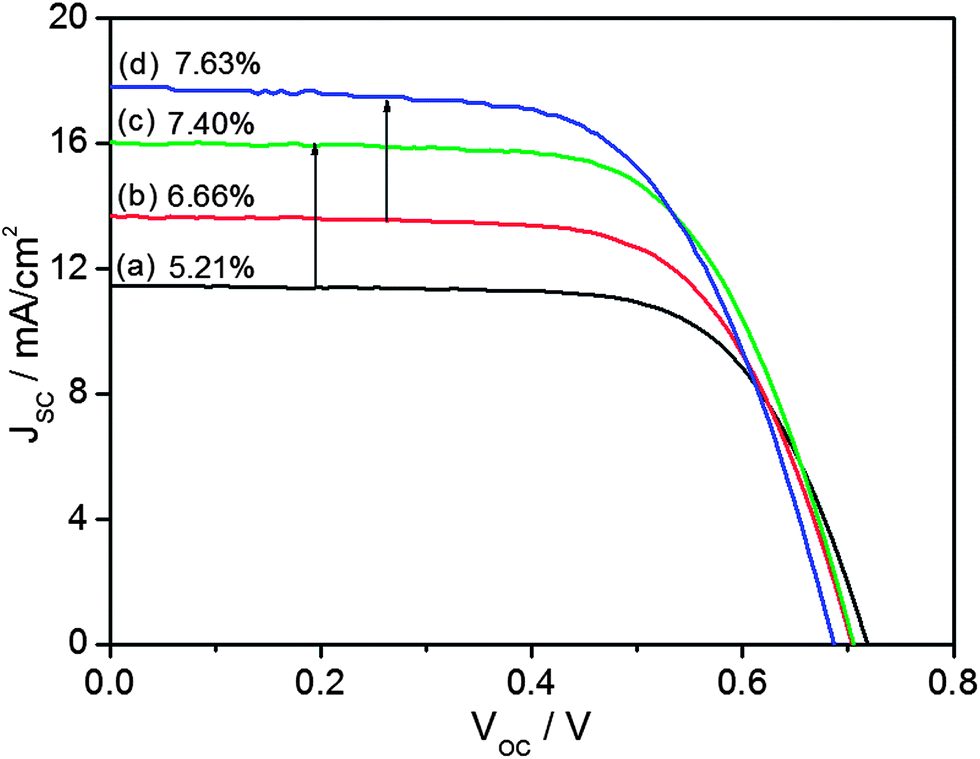 | ||
| Fig. 6 I–V curves of DSCs; (a) commercial TiO2 monolayer, (b) commercial TiO2 double layer, (c) commercial TiO2 monolayer + hollow TiO2 monolayer and (d) commercial TiO2 double layer + hollow TiO2 monolayer. | ||
Furthermore, the effect of crystallinity of hollow TiO2 NPs on conversion efficiency is also investigated. Therefore, a bilayer photoelectrode is constructed which is composed of 2.5 μm layer of the hollow TiO2 (synthesized at 180 °C for different times) on top of a 6.56 μm layer of commercial TiO2 photoelectrode. It is found that the efficiency increases from 6.21 to 7.4 with increasing reaction time from 1 h to 15 h, respectively (Fig. 7). This increase in efficiency is related to increases in crystallinity of TiO2 NPs, which is confirmed from XRD results in Fig. 1. Therefore, crystallinity also plays an important role in power conversion.
 | ||
| Fig. 7 Conversion efficiency change of DSCs with hollow TiO2 layer synthesized at 180 °C for 1–15 h. | ||
The enhanced photocurrent is most likely to be attributed to the hollow structured TiO2 scattering layer for a higher light-harvesting efficiency as well as amount of dye adsorption on the hollow TiO2 and also electrolyte diffusion feasibility.33–38 BET surface area of hollow TiO2 and commercial TiO2 was calculated by using nitrogen adsorption data in the BET region (P/Po < 0.3; where P is equilibrium and Po is saturation pressure) (ESI, Fig. S5†). The surface area of hollow TiO2 NPs and commercial TiO2 is 89.4 m2 g−1 and 82.4 m2 g−1, respectively. This higher surface area of hollow TiO2 NPs can contribute in dye adsorption, and therefore enhance the light harvesting of the cells. This is also supported from UV-visible spectra of these two samples after dye loading. It can be seen in Fig. 8a that the UV-visible (300–600 nm) absorption increases for hollow TiO2 compared to commercial TiO2, which suggest that dye loading is higher for hollow TiO2 compare to commercial TiO2. Apart from dye loading increment, a major role of the scattering layer is light back reflection and increase of light path inside the electrode. The transmittance spectra for commercial TiO2 and hollow TiO2 without dye loading are shown in Fig. 8b. It shows that the transmittance decreased at every wavelength (350–800 nm) for hollow TiO2 NPs, indicating the amount of absorbed photon prominently increased compared to commercial TiO2. Fig. 8c shows the UV-vis reflectance spectra of hollow TiO2 and commercial TiO2 NPs photoelectrode without dye loading. Both, hollow TiO2 and commercial TiO2 NPs photoelectrode, had high reflectivity in the wavelength range of 400–800 nm. However, the reflectivity of hollow TiO2 photoelectrode decreased slightly in between 400 and 500 nm. Therefore, the higher efficiency of hollow TiO2 NPs based photoelectrode indicates a strong internal light scattering within the hollow TiO2 NPs which elongated the path length of the long wavelength incident light to promote the capture of photons by the dye molecules. Apart from this, electrolyte diffusion also played an important role because the best electron transport properties resulting from the pore structure suited for the electrolyte penetration. Hollow TiO2 can provide two kinds of pores i.e. tiny internal pores were formed inside the TiO2 sphere, while large external pores were generated by the formation of interstitial voids among the spherical structures. Such inter-microsphere cavities are beneficial for the pore-filling process in DSCs and thus facilitate in the improvement of efficiency.34,44 Furthermore, the increase in efficiency with increasing crystallinity is related to gradual decrease of the energy band gap, and thereby increasing the carrier concentration.45
 | ||
| Fig. 8 (a) Absorption spectra of commercial TiO2 and hollow TiO2 with dye loading; (b) transmittance and (c) reflectance spectra of commercial TiO2 and hollow TiO2 without dye loading. | ||
Conclusions
Microwave-assisted hydrothermal method was successfully used to synthesize Au@TiO2 core–shell NPs in short time compared to conductive hydrothermal method. These Au@TiO2 core–shell NPs were composed of 40 ± 5 nm Au NPs core and 60 ± 10 nm TiO2 shell layer. TiO2 shell was composed of 17 ± 2 nm TiO2 NPs. The crystallinity of TiO2 NPs was increased with increasing synthesis temperature and well defined TiO2 NPs were formed at high synthesis temperature. The stability of Au NPs in iodide electrolyte solution was examined, which suggested that Au NPs were unstable and dissolve immediately. Therefore, the possibility of application of SPR of Au@TiO2 core–shell in DSCs was reduced because of its short term stability. Hollow TiO2 NPs (150–200 nm in diameter) were produced by selective etching of as-prepared Au@TiO2 core–shell NPs and applied as a scattering layer on top of a nanocrystalline TiO2 film, serving as the photoanode of DSCs. A high efficiency of 7.40% was achieved with TiO2 hollow spheres, compared with 5.21% for the electrode with commercial TiO2 NPs. It was found that the efficiency increased with increasing crystallinity of hollow TiO2. In addition to efficient light scattering, TiO2 hollow spheres also provided a high surface area for higher dye loading and electrolyte diffusing feasibility for better electron transport and therefore, increment in efficiency.Acknowledgements
This work was supported by (a) BK21 plus program from Ministry of Education and Human-Resource Development and (b) National Research Foundation (NRF) grant funded by the Korea government (MEST) (NRF-2010-0019626, 2012R1A2A2A01006787).Notes and references
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- C. Y. Chen, M. K. Wang, J. Y. Li, N. Pootrakulchote, L. Alibabaei, C. H. Ngoc-le, J. D. Decoppet, J. H. Tsai, C. Gratzel, C. G. Wu, S. M. Zakeeruddin and M. Gratzel, ACS Nano, 2009, 3, 3103–3109 CrossRef CAS PubMed.
- F. Gao, Y. Wang, D. Shi, J. Zhang, M. K. Wang, X. Y. Jing, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Gratzel, J. Am. Chem. Soc., 2008, 130, 10720–10728 CrossRef CAS PubMed.
- H. Qin, S. Wenger, M. Xu, F. Gao, X. Jing, P. Wang, S. M. Zakeeruddin and M. Gratzel, J. Am. Chem. Soc., 2008, 130, 9202–9203 CrossRef CAS PubMed.
- M. K. Wang, M. F. Xu, D. Shi, R. Z. Li, F. F. Gao, G. L. Zhang, Z. H. Yi, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Gratzel, Adv. Mater., 2008, 20, 4460–4463 CrossRef CAS.
- J. Chung, J. Myoung, J. Oh and S. Lim, J. Phys. Chem. C, 2010, 114, 21360–21365 CAS.
- M. F. Hossain and T. Takahashi, Mater. Chem. Phys., 2010, 124, 940–945 CrossRef CAS PubMed.
- S. Yun, J. Lee, J. Chung and S. Lim, J. Phys. Chem. Solids, 2010, 71, 1724–1731 CrossRef CAS PubMed.
- Y. Bai, Y. M. Cao, J. Zhang, M. Wang, R. Z. Li, P. Wang, S. M. Zakeeruddin and M. Gratzel, Nat. Mater., 2008, 7, 626–630 CrossRef CAS PubMed.
- S. A. Cerneaux, S. M. Zakeeruddin, M. Gratzel, Y. B. Cheng and L. Spiccia, J. Photochem. Photobiol., A, 2008, 198, 186–191 CrossRef CAS PubMed.
- S. Ito, S. M. Zakeeruddin, P. Comte, P. Liska, D. B. Kuang and M. Gratzel, Nat. Photonics, 2008, 2, 693–698 CrossRef CAS.
- T. Murakami and M. Gratzel, Inorg. Chim. Acta, 2008, 361, 572–580 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2011, 110, 6595–6663 CrossRef PubMed.
- Y. Luo, D. Li and M. Meng, Adv. Mater., 2009, 21, 4647–4651 CrossRef CAS.
- H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Mater. Chem., 2012, 22, 15475–15489 RSC.
- H. Chen, M. G. Blaber, S. D. Standridge, E. J. DeMarco, J. T. Hupp, M. A. Ratner and G. C. Schatz, J. Phys. Chem. C, 2012, 116, 10215–10221 CAS.
- Q. Zhang, D. Myers, J. Lan, S. A. Jenekhebc and G. Cao, Phys. Chem. Chem. Phys., 2012, 14, 14982–14998 RSC.
- W. Hou, P. Pavaskar, Z. Liu, J. Theiss, M. Aykol and S. B. Cronin, Energy Environ. Sci., 2011, 4, 4650–4655 CAS.
- H. Li, W. Hong, F. Cai, Q. Tang, Y. Yan, X. Hu, B. Zhao, D. Zhang and Z. Xu, J. Mater. Chem., 2012, 22, 24734–24743 RSC.
- M. D. Brown, T. Suteewong, R. S. S. Kumar, V. D'Innocenzo, A. Petrozza, M. M. Lee, U. Wiesner and H. J. Snaith, Nano Lett., 2011, 11, 438–445 CrossRef CAS PubMed.
- H. Choi, W. T. Chen and P. V. Kamat, ACS Nano, 2012, 6, 4418–4427 CrossRef CAS PubMed.
- J. Du, J. Qi, D. Wang and Z. Tang, Energy Environ. Sci., 2012, 5, 6914–6918 CAS.
- J. Qi, X. Dang, P. T. Hammond and A. M. Belcher, ACS Nano, 2011, 5, 7108–7116 CrossRef CAS PubMed.
- Q. Xu, F. Liu, W. Meng and Y. Huang, Opt. Express, 2012, 20, A898–A907 CrossRef PubMed.
- Y.-S. Kim, P. Rai and Y.-T. Yu, Sens. Actuators, B, 2013, 186, 633–639 CrossRef CAS PubMed.
- M. Giersig, T. Ung, L. M. Liz-Marzan and P. Mulvaney, Adv. Mater., 1997, 9, 570–575 CrossRef CAS.
- Z. W. Seh, S. Liu, S.-Y. Zhang, K. W. Shah and M.-Y. Han, Chem. Commun., 2011, 47, 6689–6691 RSC.
- J. Lee, J. C. Park, J. U. Bang and H. Song, Chem. Mater., 2008, 20, 5839–5844 CrossRef CAS.
- H. Yu, Y. Bai, X. Zong, F. Tang, G. Q. M. Lu and L. Wang, Chem. Commun., 2012, 48, 7386–7388 RSC.
- G. Zhu, X. Wang, H. Li, L. Pan, H. Sun, X. Liu, T. Lv and Z. Sun, Chem. Commun., 2012, 48, 958–960 RSC.
- C.-S. Chou, M.-G. Guo, K.-H. Liu and Y.-S. Chen, Appl. Energy, 2012, 92, 224–233 CrossRef CAS PubMed.
- H. Pang, H. Yang, C. X. Guo, J. Lua and C. M. Li, Chem. Commun., 2012, 48, 8832–8834 RSC.
- S. Dadgostar, F. Tajabadi and N. Taghavinia, ACS Appl. Mater. Interfaces, 2012, 4, 2964–2968 CAS.
- Y. J. Kim, M. H. Lee, H. J. Kim, G. Lim, Y. S. Choi, N.-G. Park, K. Kim and W. I. Lee, Adv. Mater., 2009, 21, 3668–3673 CrossRef CAS.
- I. G. Yu, Y. J. Kim, H. J. Kim, C. Lee and W. I. Lee, J. Mater. Chem., 2011, 21, 532–538 RSC.
- F. Huang, D. Chen, X. L. Zhang, R. A. Caruso and Y.-B. Cheng, Adv. Funct. Mater., 2010, 20, 1301–1305 CrossRef CAS.
- Y.-C. Park, Y.-J. Chang, B.-G. Kum, E.-H. Kong, J. Y. Son, Y. S. Kwon, T. Parkc and H. M. Jang, J. Mater. Chem., 2011, 21, 9582–9586 RSC.
- B. Mandlmeier, J. M. Szeifert, D. Fattakhova-Rohlfing, H. Amenitsch and T. Bein, J. Am. Chem. Soc., 2011, 133, 17274–17282 CrossRef CAS PubMed.
- P. Diao, J. Wang, D. Zhang, M. Xiang and Q. Zhang, J. Electroanal. Chem., 2009, 630, 81–90 CrossRef CAS PubMed.
- S. Singh, R. Pasricha, U. M. Bhatta, P. V. Satyam, M. Sastry and B. L. V. Prasad, J. Mater. Chem., 2007, 17, 1614–1619 RSC.
- A. Rai, A. Singh, A. Ahmad and M. Sastry, Langmuir, 2006, 22, 736–741 CrossRef CAS PubMed.
- O. M. Magnussen, Chem. Rev., 2002, 102, 679–725 CrossRef CAS PubMed.
- W. Cheng, S. Dong and E. Wang, Angew. Chem., Int. Ed., 2003, 42, 449–452 CrossRef CAS PubMed.
- W. Chen, Y. Qiu, K. Yan and S. Yang, J. Power Sources, 2011, 196, 10806–10816 CrossRef CAS PubMed.
- K.-S. Ahn, S.-H. Lee, A. C. Dillon, C. E. Tracy and R. Pitts, J. Appl. Phys., 2007, 101, 093524 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fabrication of DSCs, Crystallite size of TiO2, TEM images of Au@TiO2 core–shell NPs, Photograph of Au@TiO2 core–shell NPs and BET surface area of commercial TiO2 and hollow TiO2 NPs. See DOI: 10.1039/c3ra45860a |
| This journal is © The Royal Society of Chemistry 2014 |