Efficient adsorptive removal of dibenzothiophene by graphene oxide-based surface molecularly imprinted polymer†
Feifei
Duan
ac,
Chaoqiu
Chen
b,
Guizhen
Wang
b,
Yongzhen
Yang
ad,
Xuguang
Liu
*ac and
Yong
Qin
*b
aKey Laboratory of Interface Science and Engineering in Advanced Materials (Taiyuan University of Technology), Ministry of Education, Taiyuan 030024, China. E-mail: liuxuguang@tyut.edu.cn; Tel: +86 351-6014138
bInstitute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030024, China. E-mail: qinyong@sxicc.ac.cn; Tel: +86 351-4040081
cCollege of Chemistry and Chemical Engineering, Taiyuan University of Technology, Taiyuan 030024, China
dResearch Center on Advanced Materials Science and Technology, Taiyuan University of Technology, Taiyuan 030024, China
First published on 11th November 2013
Abstract
Molecularly imprinted polymers on GO nanosheets (MIPs/GO) for desulfurization are synthesized using dibenzothiophene (DBT) as template, methacrylic acid (MAA) as monomer and ethylene glycol dimethacrylate (EGDMA) as cross-linker. The formation of this hybrid material is verified by Fourier transform infrared spectroscopy, thermal gravimetric and atomic force microscopy analysis. The adsorption results show that the prepared MIPs/GO exhibit excellent adsorption capacity (up to 181.9 mg g−1 at 298 K) and fast mass transfer and binding kinetics for DBT. The kinetics and isotherm data can be well described by the pseudo-first-order kinetic model and the Freundlich isotherm, respectively. Competitive adsorption experiments demonstrate that MIPs/GO show higher affinity toward target molecule DBT than toward structural analogue benzothiophene.
Introduction
Sulfur-containing compounds in petroleum and in fuels are a major source of SOx emission, which results in air pollution, acid rain and catalyst poisoning issues.1,2 Therefore, deep desulfurization of fuels has attracted increasing attention in order to satisfy the ever-increasing stringent environmental regulations and fuel specifications worldwide. Various efficient methods have been developed for removal of sulfur-containing compounds from fuels, such as hydrodesulfurization (HDS),3,4 adsorptive desulfurization,5,6 oxidative desulfurization,7 extractive desulfurization8 and biodesulfurization.9,10 Among these methods, adsorptive desulfurization technology is considered as a promising method for deep desulfurization because of mild operation conditions and low impact on environment.6 More importantly, the adsorptive desulfurization can efficiently remove polyaromatic sulfur-containing compounds such as benzothiophene (BT), dibenzothiophene (DBT), and their alkyl derivatives (i.e., 4-methyldibenzothiophene and 4,6-dimethyldibenzothiophene),11 which are difficult to be removed using current HDS technology.12The key point in adsorptive desulfurization is the design of adsorbents. During the last decade, many porous materials such as active carbon,1,12 molecularly imprinted polymers (MIPs),13–16 zeolite17,18 and metal organic framework19 have been found efficient for desulfurization. Among these adsorbent materials, MIPs possess unique advantages in highly selective removal of specific polyaromatic sulfur-containing compounds with low abundance because of their molecular recognition characteristic based on their memory effect of the shape, size and functional groups of the template/target molecules.20,21 The highly selective adsorption by molecular recognition is beneficial to enrichment, separation and purification of different polyaromatic sulfur-containing compounds from fuels, which are desired considering that some complicated sulfur-containing compounds such as BT, DBT and their derivatives are important organic and medicine synthesis intermediates. Surface molecular imprinting, in which molecularly imprinted polymers are placed on the surface of supports, is very attractive for adsorptive desulfurization, because it can enhance mass transfer and binding kinetics, and hence generate higher binding capacity.21,22 Recently, MIPs were prepared on carbon microspheres using DBT as template, the adsorption capacity of DBT was 109.5 and 1.16 mg g−1 with methacrylic acid (MAA) and 2-acrylamido-2-methylpropanesulfonic acid as monomers, respectively.13,23 The carbon microspheres were chosen as supports because of their good thermal stability, excellent mechanical performance and acid/base resistance.23–26 However, the low surface area and complex, time-consuming functionalization process of carbon microspheres limit their further applications.
Graphene oxide (GO), a highly oxidized derivative of graphene, is a 2D layered nanomaterial featuring a variety of oxygen-containing functionalities with epoxide and hydroxyl groups on the basal plane and carbonyl and carboxyl groups along the edges,27,28 which provide a platform for rich chemistry to occur both within the intersheet gallery and along sheet edges29 and make the GO nanosheets more applicable for use as a support for MIPs than some inert carbon materials (such as carbon nanotubes30,31 and above mentioned carbon microspheres). Furthermore, the huge specific surface area and 2D nanosheet structure of GO are expected to supply higher adsorption capacity and faster mass transfer and adsorption kinetics.32 A few works on MIPs/GO composites for recognition of protein, dopamine and endocrine disrupting chemicals have been reported.32–34 However, to our knowledge, GO based MIPs for adsorption of sulfur-containing compounds in fuel have not been reported. In this work, we report on the synthesis of surface molecularly imprinted polymers on GO for deep desulfurization using DBT as template molecule, MAA as functional monomer and ethylene glycol dimethacrylate (EGDMA) as cross-linker. Adsorption results clearly demonstrate that the MIPs/GO exhibit excellent adsorption capacity and removal efficiency for BDT. Model fitting of the isotherms indicates that the adsorption imply a Freundlich isotherm behavior. The maximum adsorption capacity of MIPs/GO toward DBT (up to 181.9 mg g−1) is twice higher than that of GO (97 mg g−1). Competitive adsorption experiments show that MIPs/GO exhibit higher affinity toward DBT than toward structural analogue benzothiophene.
Experimental section
Synthesis of GO
GO was prepared from natural graphite flakes using a modified Hummers method35 through acid oxidation of flake graphite. In brief, flake graphite (2 g) was put into cold (0 °C) concentrated H2SO4 (46 mL) solution in a 1 L flask. While maintaining vigorous stirring, 6 g of KMnO4 was slowly added to the flask and the temperature was kept below 20 °C. The mixture was stirred at 35 °C for 1 h until it became pasty brownish, and then deionized water (92 mL) was added dropwise under ice-bath. After that, the mixture was stirred at 90 °C for 1 h, and then diluted with deionized water (280 mL). After vigorous stirring for 2 h, 50 mL of 30 wt% H2O2 solution was slowly added into the mixture, then the colour of the mixture changed to bright yellow. The mixture was filtered and washed with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 HCl solution for several times to remove the residual metal ions. The resulting solid was suspended in water under ultrasonication for 5 h, followed by centrifugation at 4000 rpm for 0.5 h. The obtained supernate was dried in air at 80 °C, yielding GO sample.
:10 HCl solution for several times to remove the residual metal ions. The resulting solid was suspended in water under ultrasonication for 5 h, followed by centrifugation at 4000 rpm for 0.5 h. The obtained supernate was dried in air at 80 °C, yielding GO sample.
Preparation of adsorbent
GO was first modified by a polymerizable silane coupling agent 3-methacryloxypropyl trimethoxysilane (MPS). Typically, 2 mL of MPS and 15 mL of distilled water were added to 30 mL of acetonitrile with 0.3 g of dispersed GO. The mixture was stirred for 4 h at 65 °C. After the reaction, the product was separated by centrifugation, washed with ethanol and dried at 50 °C overnight to obtain MPS/GO. Then 0.369 g of DBT and 1 mL of MAA were dissolved in 50 mL of acetonitrile with dispersed 0.2 g MPS/GO. After assembly for 1 h, 4 mL of EGDMA and 0.065 g of 2, 2′-azobisisobutyronitrile (AIBN) were added into the mixture. The suspension was sonicated for 10 min and deoxygenized with nitrogen for 1 h. The polymerization was performed at 50 °C for 24 h under nitrogen. The product (denoted as MIPs-DBT/GO) was centrifuged and washed with 30 mL of mixture solution (ethanol and acetic acid, v/v = 9/1) under ultrasound for five times to remove unreacted reagents and template molecules (DBT). Finally, the product was washed with ethanol to remove the remaining acetic acid and dried under vacuum. The product was denoted as MIPs/GO. For comparison, the surface nonimprinted polymers (NIPs/GO) were also prepared in the same way without adding template molecules (DBT).Adsorption test
Adsorption of DBT was carried out in a stirred batch system at different temperature. In brief, DBT was dissolved in n-hexane as a stock solution (2000 mg L−1) and further diluted with a certain amount of acetonitrile to the required concentration before being used. All adsorption experiments were performed in sealed 100 mL glass conical bottles that contained 20 mg of adsorbent and 50 mL of DBT solution in the appropriate concentration. The suspension was ultrasonicated for 15 minutes with carefully controlled temperature, and then placed in a water bath at an appropriate temperature. The mixture was sampled periodically for analysis by gas chromatography (SHIMADZU GC2014C) fitted with a DB-5 capillary column (30 m × 0.32 mm) and flame ionization detector.The adsorption kinetics study under different temperature was carried out with an initial DBT concentration of 500 mg L−1 to determine the minimum time required for adsorption to reach equilibrium and the temperature for adsorption to reach the maximum adsorption capacity. The concentration of DBT was determined at different time intervals ranging from 5 minutes to 1 h. The adsorption isothermal experiments under different initial DBT concentration were similar to the adsorption kinetics study. The initial DBT concentration ranged from 10 mg L−1 to 500 mg L−1. The competitive adsorption test was performed in a mixture solution of DBT and BT, and the initial concentrations of BT and DBT were both 500 mg L−1.
The adsorption capacity of adsorbent (mg g−1) at equilibrium qe was calculated according to the following formula:
 | (1) |
Characterization
The morphologies of the samples were characterized by transmission electron microscopy (TEM, JEOL-2100F). Atomic force microscopy (AFM) images were recorded on an SPI 3800N atomic force microscope (in tapping mode). Fourier-transform IR (FTIR) spectra were recorded on a Bruker Tensor 27 instrument. The thermal stability of the samples was analyzed by thermogravimetry (TG) using a Netzsch TG209 F3 instrument at a heating rate of 5 °C min−1 under N2 atmosphere.Results and discussion
Preparation and characterization of MIPs/GO hybrids
Here, GO was chosen as support for MIPs since its very high surface area and selective adsorption ability toward the aromatic compounds with benzene rings through strong π–π interaction36 make itself a potential adsorbent for desulfurization. The MIPs/GO hybrid was prepared through a molecular imprinting route, as shown in Scheme 1. Firstly, the silane coupling agent MPS was grafted onto GO for introducing vinyl groups, which supplied reacting sites for imprinted polymerization. Subsequently, template–monomer complexes and crosslink agent (EGDMA) were added into the system, formed a thin layer of imprinted polymers on GO. After removal of the template molecules (DBT) using acid washing to destroy the hydrogen bonds between monomer and template, recognition cavities complementary to the DBT in shape, size and chemical functionality were formed in the MIPs/GO hybrids, which could selectively rebind DBT molecules from a mixture of closely related compounds. | ||
| Scheme 1 Scheme of molecular imprinting process on GO surface. | ||
FTIR analysis was carried out to characterize the structure details of the GO, MPS/GO, MIPs-DBT/GO and MIPs/GO samples. In the FTIR spectrum of GO (Fig. 1A), the strong bands at 3413 cm−1 and 1725 cm−1 correspond to the –OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, respectively, while the band at 1621 cm−1 indicates the existence of aromatic CC groups.36 The bands at around 1383 cm−1 and 1131 cm−1 can be assigned to carboxyl OC–O bonds37 and C–O groups, respectively. Compared with GO, MPS/GO shows a new band at 1038 cm−1, which can be attributed to Si–O groups.33 While the expected CC bands in MPS is overlapped with the characteristic peaks of aromatic CC groups of GO. These bands indicate MPS is successfully introduced onto the GO. The FTIR spectrum of MIPs-DBT/GO and MIPs/GO also clearly show the characteristic Si–O and CO stretching vibrations of carboxylic ester centered at 1725 cm−1 and 1038 cm−1, respectively. The bands at 3051 cm−1 and 1366 cm−1 in MIPs-DBT/GO can be assigned to C–H in benzene and thiophene ring, which are the characteristic bands of the template DBT molecules. After washing with ethanol and acetic acid for five times, no characteristic bands of DBT can be observed in the FTIR spectra of MIPs/GO, indicating that the template DBT molecules were removed from MIPs/GO composite.
O groups, respectively, while the band at 1621 cm−1 indicates the existence of aromatic CC groups.36 The bands at around 1383 cm−1 and 1131 cm−1 can be assigned to carboxyl OC–O bonds37 and C–O groups, respectively. Compared with GO, MPS/GO shows a new band at 1038 cm−1, which can be attributed to Si–O groups.33 While the expected CC bands in MPS is overlapped with the characteristic peaks of aromatic CC groups of GO. These bands indicate MPS is successfully introduced onto the GO. The FTIR spectrum of MIPs-DBT/GO and MIPs/GO also clearly show the characteristic Si–O and CO stretching vibrations of carboxylic ester centered at 1725 cm−1 and 1038 cm−1, respectively. The bands at 3051 cm−1 and 1366 cm−1 in MIPs-DBT/GO can be assigned to C–H in benzene and thiophene ring, which are the characteristic bands of the template DBT molecules. After washing with ethanol and acetic acid for five times, no characteristic bands of DBT can be observed in the FTIR spectra of MIPs/GO, indicating that the template DBT molecules were removed from MIPs/GO composite.
 | ||
| Fig. 1 (A) FTIR spectra of GO, MPS/GO, MIPs-DBT/GO, MIPs/GO and DBT, (B) TG curves of GO, MPS/GO and MIPs/GO. | ||
GO, MPS/GO and MIPs/GO were further studied using TG technique to get more information about the functional groups on the surface of GO. The TG curves for these three samples are shown in Fig. 1B. The weight of GO declines sharply between 100 °C and 200 °C, which can be attributed to desorption of the adsorbed water molecules and removal of oxygenated functional groups from the GO.32 The weight loss of GO is 28.77% obtained from the TG analysis. Compared with GO, MPS/GO and MIPs/GO show faster loss between 100 °C and 200 °C because of the thermal decomposition of MPS and polymer. Moreover, at 252 °C (the temperature of the final decomposition determined by DTG, Fig. S1†) the weight loss of MPS/GO and MIPs/GO are 33.49% and 38.64%, respectively. Because the residue of MPS itself is 61.5%,23 the graft content of MPS/GO (x) is 14.42%, calculated as follow: x × 61.5% + (1 − x) × 28.77% = 33.49%. This result is consistent with that of TG analysis under air atmosphere (Fig. S2†). For MIPs/GO, the residue of MIPs is hard to measure, so we calculated the graft content without considering the residue of MIPs. The graft content of MIPs/GO is 7.74% calculated based on MPS/GO as follow: x + (1 − x) × 33.49% = 38.64%. These difference in thermal stability between GO, MPS/GO and MIPs/GO shows that molecularly imprinted polymers are successfully grafted on the GO surface.
The thicknesses of GO sheets and MIPs/GO were probed by AFM. The thickness of GO as measured from the cross section analysis is on the order of 3 nm, corresponding to double-layer nanosheets (Fig. 2A). Similar results were also observed in other AFM studies.38,39 According to AFM image of MIPs/GO, the average thickness of the polymers grafted onto the GO surface is about 22 nm (Fig. 2B). The morphological structure of GO and MIPs/GO was examined by TEM. From the TEM images (Fig. 2C and D), it can be seen that the GO is very thin and contains some wrinkles. These wrinkles are considered to be important for preventing aggregation of GO and maintaining high surface area.40 For MIPs/GO, it can be observed that the GO sheets are mainly decorated by the MIPs film, indicating that GO nanosheets are suitable supports for molecular imprinting process. The AFM and TEM results further confirm that the molecularly imprinted polymers are grafted on the GO nanosheets.
 | ||
| Fig. 2 AFM images and cross section views (A and B) and TEM images (C and D) of GO and MIPs/GO. | ||
Adsorption tests
To evaluate the effectiveness of MIPs for adsorption of DBT, the adsorption capacity and equilibrium time of GO, MIPs/GO and NIPs/GO were investigated at 298 K with initial DBT concentration of 500 mg L−1. Fig. 3 displays the adsorption kinetics curves of GO, MIPs/GO and NIPs/GO for DBT. It can be seen clearly that the equilibrium time is about 35 minutes for all samples, which is faster than our previous results for MIPs-carbon microspheres (about 300 min),13 indicative of quite fast mass transfer on these GO-based materials because of the unique 2D nanostructure. As expected, the adsorption capacity of MIPs/GO (181.9 mg g−1) is much higher than NIPs/GO (106.3 mg g−1) and GO (97 mg g−1), demonstrating the formation of specific recognition site on the surface of GO, which benefits DBT to bind with the recognition sites. The adsorption capacity of MIPs/GO is also much higher than our previous MIPs-carbon microspheres (Table 3), which can be attributed to the larger surface areas of the GO nanosheets. | ||
| Fig. 3 Effect of contact time on the adsorption of DBT by GO, MIPs/GO and NIPs/GO (20 mg of samples in 50 mL of DBT solution at 298 K). | ||
To investigate the mechanism of the adsorption process of MIPs/GO for DBT, adsorption kinetics tests at different temperatures (293 K, 298 K and 303 K) were carried out and two conventional kinetic models (pseudo-first-order and pseudo-second-order) were applied to analyse the experimental data.
The pseudo-first-order model can be described as:41,42
| ln(qe − qt) = lnqe − k1t | (2) |
The pseudo-second-order model comprises all the steps of adsorption including external film diffusion, adsorption, and internal particle diffusion, which is described as:43
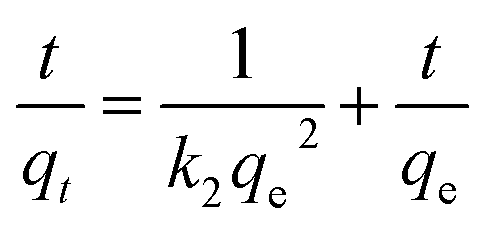 | (3) |
A comparison of the kinetic models for removal of DBT by MIPs/GO using nonlinear regression is presented in Fig. 4, and the corresponding kinetic parameters and correlation coefficients are summarized in Table 1. As shown in Fig. 4 and Table 1, the pseudo-first-order model is better than pseudo-second-order model in describing adsorption of DBT on MIPs/GO under all three temperatures according to the higher correlation coefficient values. Moreover, the experimental adsorption capacity (qe,exp) is also in accordance with the calculated adsorption capacity (qe,cal) obtained from pseudo-first-order model. These results indicate that the pseudo-first-order kinetic model fit the adsorption of DBT on MIPs/GO better than the pseudo-second-order model.
 | ||
| Fig. 4 Adsorption kinetics of DBT on MIPs/GO at three different temperatures (20 mg of MIPs/GO in 50 mL of DBT solution for 1 h). The solid line is pseudo-first-order model simulation; the dash line is pseudo-second-order model simulation. | ||
| Temperature | q e,exp (mg g−1) | Pseudo-first-order | Pseudo-second-order | ||||
|---|---|---|---|---|---|---|---|
| K 1 (min−1) | q e,cal (mg g−1) | R 2 | K 2 (min−1) | q e,cal (mg g−1) | R 2 | ||
| 293 K | 94.9 | 0.0689 | 98.23 | 0.9871 | 0.00052 | 101.43 | 0.9599 |
| 298 K | 181.9 | 0.07662 | 189.61 | 0.9909 | 0.00033 | 194.34 | 0.9635 |
| 303 K | 59.7 | 0.06906 | 62.59 | 0.9858 | 0.0008 | 64.79 | 0.9424 |
The adsorption isotherms of DBT on MIPs/GO at four different temperatures (Fig. 5) are given to investigate the interaction between adsorbent and adsorbate when the adsorption process reaches equilibrium. It can be observed that the adsorption capacity of MIPs/GO increases with the increasing equilibrium concentration of DBT. This can be attributed to the increasing driving force of the concentration gradient because the increase in DBT concentration can accelerate the diffusion of DBT molecules onto MIPs/GO.36 Langmuir model and Freundlich model are used to describe the adsorption isotherms of DBT on MIPs/GO. The Langmuir model assumes that the adsorption takes place on a homogeneous surface with monolayer coverage and uniform energies. The Freundlich model is an empirical model based on multilayer adsorption on heterogeneous surfaces with the exponential distribution of active sites and energies.14
 | ||
| Fig. 5 Adsorption isotherms of DBT by MIPs/GO at four different temperatures (20 mg of MIPs/GO in 50 mL of DBT solution for 1 h). The solid line is Freundlich model simulation; the dash line is Langmuir model simulation. | ||
The equation of Langmuir model can be described as:44
 | (4) |
The equation of Freundlich model is expressed as:45,46
 | (5) |
qeversus lnCe.
A comparison of the isotherm models for DBT adsorption on MIPs/GO using nonlinear regression is also given in Fig. 5, and the parameters calculated from isotherm models using nonlinear regression are listed in Table 2. The correlation coefficient R2 values of the Freundlich model at four different temperatures exceeded 0.99, which is much higher than those of the Langmuir model. Moreover, the values of Freundlich constant n are all greater than 1 at four temperatures, representing the favorable adsorption conditions,47 and the values of Freundlich constant KF indicate that the adsorption capacity initially increases and subsequently decreases from 273 K to 303 K, which agrees with experimental data. The results indicate Freundlich model is more suitable to describe the adsorption isotherm of DBT on MIPs/GO and the adsorption occurred in a multilayer adsorption manner.14 From 273 K to 298 K, the adsorption capacity is increased with increasing temperature, suggesting the endothermic nature of adsorption. However, the adsorption capacity at 303 K is lower than that at 298 K, maybe because of the breakage of hydrogen bonds between functional monomer and template with increasing temperature, resulting in the decrease of adsorption capacity.
| Temperature | Langmuir | Freundlich | ||||
|---|---|---|---|---|---|---|
| q m (mg g−1) | K L (L mg−1) | R 2 | K F | n | R 2 | |
| 273 K | 79.99 | 0.0002 | 0.9688 | 0.0197 | 1.1341 | 0.9994 |
| 293 K | 96.624 | 0.00002 | 0.9735 | 0.03637 | 1.2975 | 0.9962 |
| 298 K | 176.645 | 0.00003 | 0.9924 | 0.1808 | 1.1413 | 0.9996 |
| 303 K | 55.578 | 0.00002 | 0.9734 | 0.0105 | 1.4271 | 0.9986 |
Table 3 compares the adsorption capacity of MIPs/GO for DBT against other surface molecularly imprinted polymers previously reported in the literature. Apparently, the adsorption capacity of MIPs/GO for DBT is higher. Moreover, the adsorption equilibrium time of DBT on MIPs/GO is much shorter, which can be due to the more accessible recognition sites on the platform of GO.32 These results confirm that the MIPs/GO can be used as a superior material for adsorptive desulfurization.
| Support | Functional monomer | Temperature (K) | Equilibrium time (min) | Adsorption capacity (mg g−1) | Ref. |
|---|---|---|---|---|---|
| GO | MAA | 298 | 35 | 181.9 | This work |
| Nano-TiO2 as sacrificial support | 4-Vinylpyridine | 318 | 240 | 26.74 | 15 |
| Silica gel | MAA | Room temperature | 120 | 57.4 | 48 |
| Carbon microspheres | MAA | Room temperature | 300 | 109.5 | 13 |
| Potassium hexatitanate whisker | 4-Vinylpyridine | 318 | 240 | 22.03 | 14 |
To evaluate the specificity of MIPs/GO for DBT, competitive adsorption experiments were carried out using BT as the reference compound. The selectivity adsorption capacity of GO, MIPs/GO and NIPs/GO are shown in Fig. 6. It can be observed that the adsorption capacity of GO toward BT and DBT are 68.8 mg g−1 and 26.4 mg g−1, respectively. For NIPs/GO, the adsorption capacities toward DBT and BT in the binary mixture system have no significant differences. In contrast, the adsorption capacity of MIPs/GO toward BT and DBT are 74.7 mg g−1 and 113 mg g−1, respectively, and the adsorption coefficient is 1.57. BT is smaller than DBT, allowing its easy accessibility to the surface of GO. However, it is not matching with the imprinted cavities and spatial arrangement of imprinted sites in MIPs/GO. In NIPs/GO, there are no imprinted cavities and sites generated during polymerization, resulting in nonspecific interaction with DBT and BT. These results confirm that MIPs/GO have excellent recognition ability and high selectivity for DBT in the presence of the structural analogues.
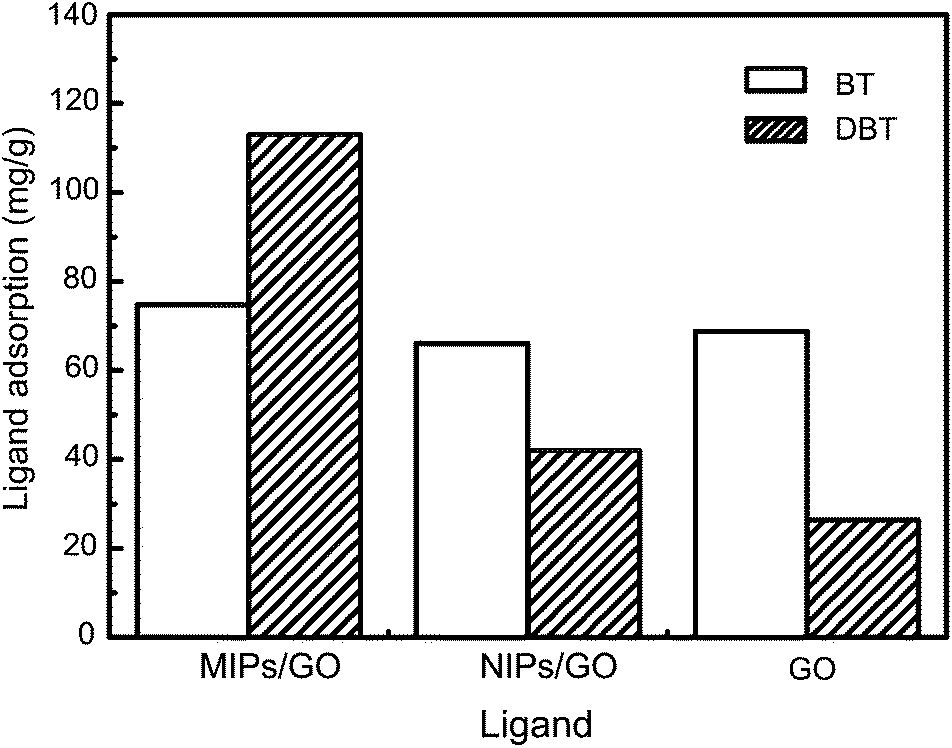 | ||
| Fig. 6 Competitive adsorption of DBT with BT onto GO, MIPs/GO and NIPs/GO. | ||
Conclusion
In summary, molecularly imprinted polymers onto GO nanosheets were fabricated for deep desulfurization using DBT as template molecule, MAA as functional monomer and EGDMA as cross-linker. The MIPs/GO showed excellent adsorption capacity (up to 181.9 mg g−1 at 298 K), high selectivity and fast mass transfer and binding kinetics for DBT, indicating GO nanosheets are suitable supports for MIPs because of their high surface area and 2D layer structure with a variety of oxygen-containing groups. The kinetics and isotherm data well fitted with the pseudo-first-order kinetic model and the Freundlich isotherm, respectively. This work opens a new possibility for development of GO-based molecularly imprinted polymer materials for the desulfurization of fuel.Acknowledgements
The authors acknowledge financial support from Program for Changjiang Scholar and Innovative Research Team in University (IRT0972), National Natural Science Foundation of China (20971094, 21176169, 51152001), Ph.D. Programs Foundation of Ministry of Education of China (20101402110007), International S&T Co-operation Program of Shanxi Province (2010081017), Research Project Supported by Shanxi Scholarship Council of China (2012-038), the Hundred Talent Program of the Chinese Academy of Sciences.Notes and references
- C. O. Ania and T. J. Bandosz, Langmuir, 2005, 21, 7752 CrossRef CAS PubMed.
- J. Xiao, C. Song and X. Ma, Ind. Eng. Chem. Res., 2012, 51, 3436 CrossRef CAS.
- D. Solís, A. L. Agudo and J. Ramírez, Catal. Today, 2006, 116, 469 CrossRef PubMed.
- A. Infantes-Molina, A. Romero-Pérez and V. Sánchez-González, ACS Catal., 2011, 1, 175 CrossRef CAS.
- C. Yu, X. Fan and L. Yu, Energy Fuels, 2013, 27, 1499 CrossRef CAS.
- Y. Shen, P. Li and X. Xu, RSC Adv., 2012, 2, 1700 RSC.
- J. Zhang, A. Wang and X. Li, J. Catal., 2011, 279, 269 CrossRef CAS PubMed.
- C. Asumana, G. Yu and X. Li, Green Chem., 2010, 12, 2030 RSC.
- F. Davoodi-Dehaghani, M. Vosoughi and A. A. Ziaee, Bioresour. Technol., 2010, 101, 1102 CrossRef CAS PubMed.
- M. A. Dinamarca, C. Ibacache-Quiroga and P. Baeza, Bioresour. Technol., 2010, 101, 2375 CrossRef CAS PubMed.
- M. Seredych and T. J. Bandosz, Energy Fuels, 2010, 24, 3352 CrossRef CAS.
- M. Seredych, J. Rawlins and T. J. Bandosz, Ind. Eng. Chem. Res., 2011, 50, 14097 CrossRef CAS.
- Y. Yang, X. Liu and M. Guo, Colloids Surf., A, 2011, 377, 379 CrossRef CAS PubMed.
- W. Yang, W. Zhou and W. Xu, J. Chem. Eng. Data, 2012, 57, 1713 CrossRef CAS.
- W.-z. Xu, W. Zhou and P.-p. Xu, Chem. Eng. J., 2011, 172, 191 CrossRef CAS PubMed.
- P. Xu, W. Xu and X. Zhang, Microchim. Acta, 2010, 171, 441 CrossRef CAS.
- T. Isoda, Y. Takase and K. Kusakabe, Energy Fuels, 2000, 14, 585 CrossRef CAS.
- L. Lin, A. Wang and M. Dong, J. Hazard. Mater., 2012, 203–204, 204 CrossRef CAS PubMed.
- D. Peralta, G. Chaplais and A. Simon-Masseron, Energy Fuels, 2012, 26, 4953 CrossRef CAS.
- X. Shen, L. Zhu and N. Wang, Chem. Commun., 2012, 48, 788 RSC.
- L. Chen, S. Xu and J. Li, Chem. Soc. Rev., 2011, 40, 2922 RSC.
- C. Xie, Z. Zhang and D. Wang, Anal. Chem., 2006, 78, 8339 CrossRef CAS PubMed.
- Y. Yang, Y. Zhang and S. Li, Appl. Surf. Sci., 2012, 258, 6441 CrossRef CAS PubMed.
- Y. Wang, F. Su and J. Y. Lee, Chem. Mater., 2006, 18, 1347 CrossRef CAS.
- Y. Z. Jin, C. Gao and W. K. Hsu, Carbon, 2005, 43, 1944 CrossRef CAS PubMed.
- Z. Wen, Q. Wang and Q. Zhang, Electrochem. Commun., 2007, 9, 1867 CrossRef CAS PubMed.
- L. Sun, H. Yu and B. Fugetsu, J. Hazard. Mater., 2012, 203–204, 101 CrossRef CAS PubMed.
- O. C. Compton, B. Jain and D. A. Dikin, ACS Nano, 2011, 5, 4380 CrossRef CAS PubMed.
- O. C. Compton, D. A. Dikin and K. W. Putz, Adv. Mater., 2010, 22, 892 CrossRef CAS PubMed.
- T. Morishita, M. Matsushita and Y. Katagiri, Carbon, 2010, 48, 2308 CrossRef CAS PubMed.
- R. Gao, X. Su and X. He, Talanta, 2011, 83, 757 CrossRef CAS PubMed.
- Y. Li, X. Li and C. Dong, Carbon, 2010, 48, 3427 CrossRef CAS PubMed.
- Y. Zeng, Y. Zhou and L. Kong, Biosens. Bioelectron., 2013, 45, 25 CrossRef CAS PubMed.
- J. Luo, S. Jiang and X. Liu, J. Phys. Chem. C, 2013, 117, 18448 CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- J. Xu, L. Wang and Y. Zhu, Langmuir, 2012, 28, 8418 CrossRef CAS PubMed.
- V. Chandra, J. Park and Y. Chun, ACS Nano, 2010, 4, 3979 CrossRef CAS PubMed.
- J. Paredes, S. Villar-Rodil and A. Martinez-Alonso, Langmuir, 2008, 24, 10560 CrossRef CAS PubMed.
- L. J. Cote, F. Kim and J. Huang, J. Am. Chem. Soc., 2009, 131, 1043 CrossRef CAS PubMed.
- H. C. Schniepp, J.-L. Li and M. J. McAllister, J. Phys. Chem. B, 2006, 110, 8535 CrossRef CAS PubMed.
- M. Li, H. Wang and S. Wu, RSC Adv., 2012, 2, 900 RSC.
- S. Deng, R. Wang and H. Xu, J. Mater. Chem., 2012, 22, 10055 RSC.
- A. S. Bhatt, P. L. Sakaria and M. Vasudevan, RSC Adv., 2012, 2, 8663 RSC.
- Y. Yuan, F. Sun and H. Ren, J. Mater. Chem., 2011, 21, 13498 RSC.
- R. J. Umpleby, S. C. Baxter and Y. Chen, Anal. Chem., 2001, 73, 4584 CrossRef CAS.
- Z. Yan, G. Li and L. Mu, J. Mater. Chem., 2006, 16, 1717 RSC.
- H. Deng, L. Gao and S. Zhang, Ind. Eng. Chem. Res., 2012, 51, 14018 CrossRef CAS.
- T.-p. Hu, Y.-m. Zhang and L.-h. Zheng, J. Fuel Chem. Technol., 2010, 38, 722 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 andS2. See DOI: 10.1039/c3ra45354e |
| This journal is © The Royal Society of Chemistry 2014 |