Stereoselective total synthesis of (+)-hyptolide†
Gowravaram
Sabitha
*,
A.
Raju
,
C. Nagendra
Reddy
and
J. S.
Yadav
Natural Products Chemistry Division, CSIR-Indian Institute of Chemical Technology (IICT), Hyderabad 500 007, India. E-mail: gowravaramsr@yahoo.com; sabitha@iict.res.in; Fax: +91-40-27160512
First published on 5th November 2013
Abstract
Stereoselective synthesis of the naturally occurring 6-membered lactone hyptolide 1 is described. The main feature of the synthesis is the utility of the hitherto unexplored alkyne fragment derived from commercially available 3-butyn-1-ol (homopropargylic alcohol).
Introduction
In 1920, Gorter1 reported the isolation of a crystalline lactone, (+)-hyptolide 1, from leaves of Hyptis pectinata, a plant that is used in folk medicine. Various biological effects were found with its extracts, such as: analgesic, anti-inflammatory, and anti-microbial. The structure proposed was proved wrong when hyptolide was subjected to reinvestigation by Birch and Butler2 in 1964. Later, the absolute configuration was established by Anders Kjaer et al.3 by detailed 1H and 13C NMR spectroscopic studies and on the basis of single-crystal X-ray diffraction data as 6R-(1Z,3S,5R,6S)-5,6-dihydro-6-[3,5,6-tris(acetoxy)-1-heptenyl]-2H-pyran-2-one. Hyptolide (1) is structurally similar to other members of the polyhydroxy δ-pyranone natural product family such as spicigerolide (2),4 synrotolide (3),5 synargentolide A (4),6 anamarine (5)7 (Fig. 1). Earlier, two syntheses8 were reported for the synthesis of (+)-hyptolide. While our manuscript was under preparation, a silicon tethered RCM route for hyptolide was published by Kumar and his co-workers.9 As part of our continuing interest in the synthesis of natural products having δ-lactone rings,10 we were interested in the synthesis of hyptolide. In the present communication, we disclose the stereoselective total synthesis of (+)-hyptolide from commercially available 3-butyn-1-ol.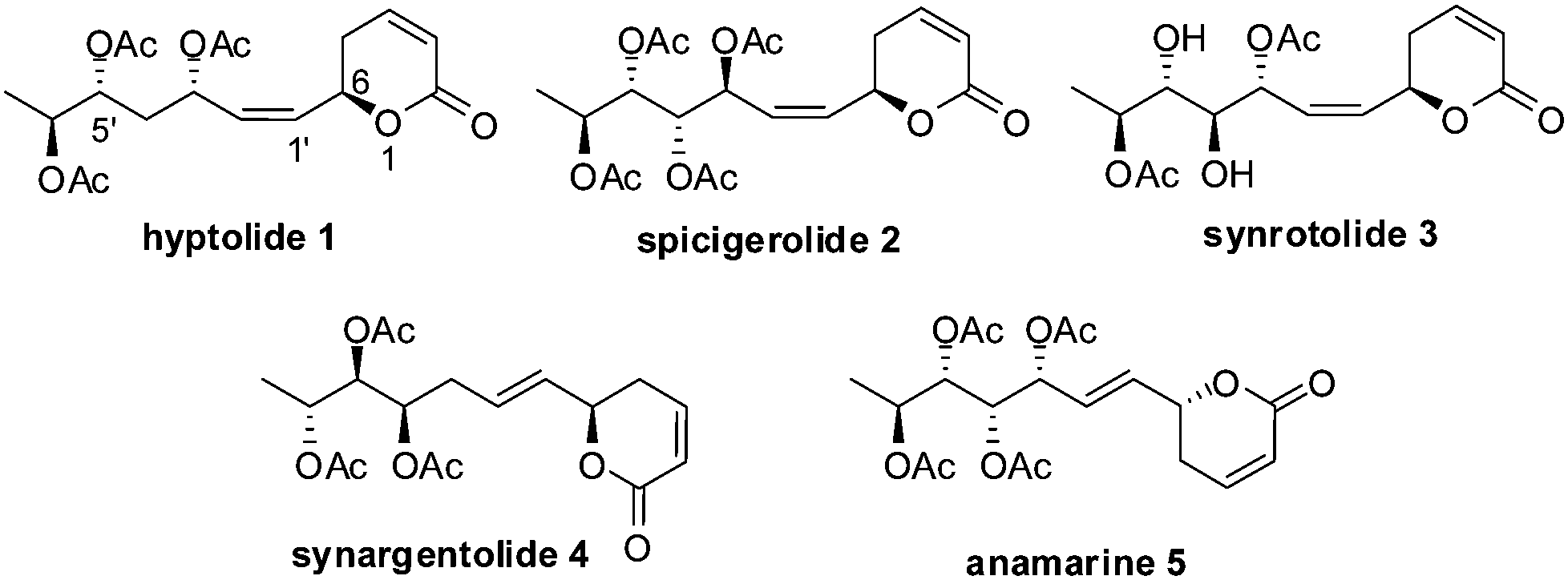 | ||
| Fig. 1 Hyptolide type pyranone polyacetates. | ||
Retrosynthetic analysis (Scheme 1) reveals that compound 1 could be synthesized from homoallylic alcohol 6 by acryloylation followed by a RCM reaction and semihydrogenation of the triple bond. The compound 6 in turn can be synthesized from 7 and 8 by a coupling reaction. Whereas, the aldehyde 7 and alkyne 8 intermediates could be derived from 3-butyn-1-ol.
 | ||
| Scheme 1 Retrosynthesis. | ||
Results and discussion
Our synthetic endeavor began with the benzyl protected 2,3-epoxy alcohol 9 (Scheme 2) prepared from 3-butyn-1-ol similarly as reported for the preparation of PMB(para-methoxybenzyl) protected 2,3-epoxy alcohol.11 We then turned our attention to the key step for the introduction of benzoate via nucleophilic epoxide ring opening with benzoic acid, which culminates in the creation of a C5′ chiral center of the molecule. Thus, regio- and stereoselective ring opening of ((2S,3S)-3-(2-(benzyloxy)ethyl)oxiran-2-yl)methanol 9 was achieved at C-3 with benzoic acid and Ti(OiPr)4 within 1 h in CH2Cl2,12 affording a 75% yield of the benzoate diol 10. The diol 10 was converted into epoxide 11 in 90% yield by following the Forsyth protocol.13 Lithium aluminium hydride reduction of epoxide 11 delivered terminal methyl compound 12. The diol 12 having anti-configuration was protected as an acetonide 13 followed by removal of benzyl group resulted in primary alcohol 14. Swern oxidation of 14 led to the formation of aldehyde 7. | ||
| Scheme 2 Synthesis of aldehyde 7. | ||
The synthesis of hitherto unexplored alkyne fragment 8 began by converting commercially available 3-butyn-1-ol (homopropargylic alcohol) to the known PMB protected chiral propargylic alcohol 15 following a reported procedure (Scheme 3).10g,14a–c The free secondary hydroxyl group of the latter was protected as silyl ether 16 and DDQ-mediated oxidative cleavage of the PMB (para-methoxybenzyl) ether produced primary alcohol 17. Swern oxidation of alcohol afforded aldehyde followed by one-carbon Wittig olefination (n-BuLi, CH3PPh3+I−, dry THF) to furnish key intermediate 8.
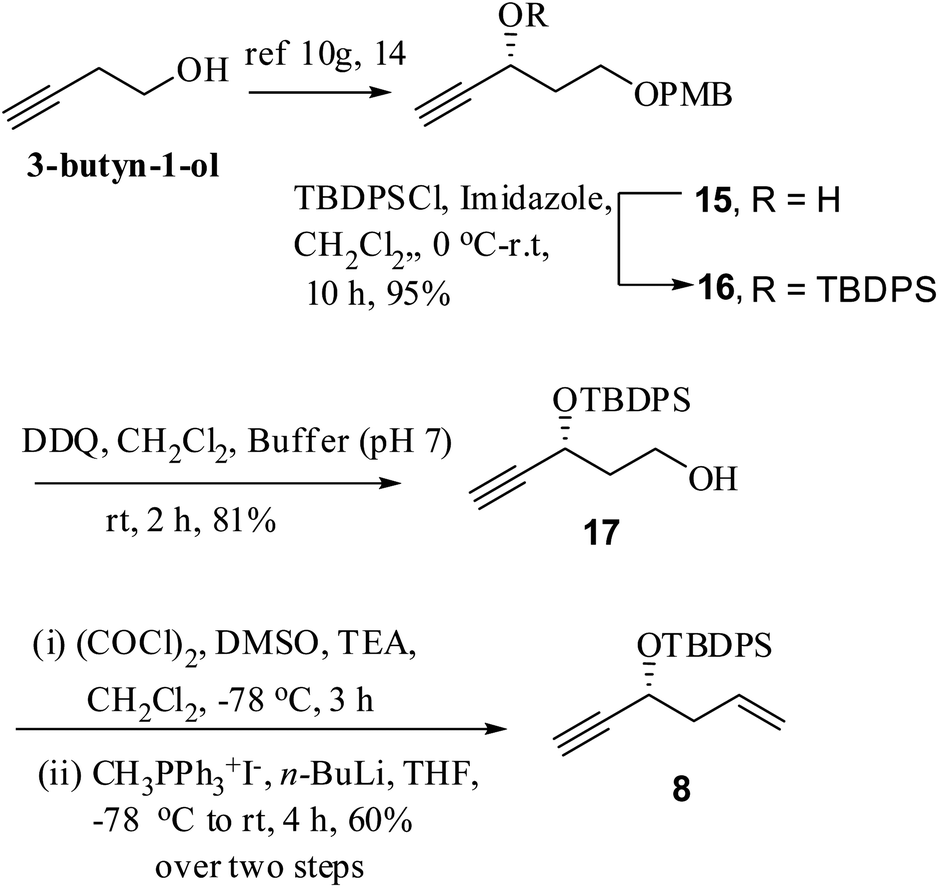 | ||
| Scheme 3 Synthesis of chiral propargylic alcohol 8. | ||
At this point, we were set to couple the appropriate fragments to generate the carbon frame work of hyptolide. Formation of the chiral propargyl alcohol 21 was envisioned by lithiated alkyne 8 coupling to the aldehyde 7 followed by adopting an oxidation/selective reduction protocol. Alkyne 8 on reaction with n-BuLi followed by coupling of the generated lithium acetylide with aldehyde 7 afforded a mixture of propargylic alcohols 18 (65%), which on oxidation with Dess–Martin Periodinane (DMP) gave ynone 19 in 82% yield (Scheme 4).
 | ||
| Scheme 4 Synthesis of hyptolide 1. | ||
Initially, our efforts towards the reduction of 19 to the corresponding hydroxy compound 21 were not fruitful under CBS conditions. Use of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M solution of BH3·DMS at −40 °C gave low yields of the reduced product. Whereas, using conc. BH3·DMS solution either in dry toluene or THF furnished the alcohol (21) as a diastereomeric mixture in 6:4 ratio however, the ketone functionality was reduced with Noyori's catalyst Ru[(1S,2S)-p-TsNCH(Ph)CH(Ph)NH](η6-p-cymene) (20)15 in HCOOH affording 21 in 90% yield and 95% de.16 Then, we tried to minimize the use of protecting groups by maintaining the oxygenated functional groups as acetates. Thus, hydrolytic cleavage of acetonide protecting group in 21 leading to a triol, which was acetylated in situ with acetic anhydride in the presence of NEt3 and DMAP to form the tri-acetate 22 in 90% yield for the two steps. The alcohol 22 was transformed into its acrylic ester 23 in 80% yield by treating with acryloyl chloride, catalytic amounts of DMAP and triethylamine in CH2Cl2. Ring closing metathesis17 reaction of 23 in refluxing DCM for 24 h using 10 mol% first generation Grubbs' catalyst, bis(tricyclohexylphosphine)benzylidene ruthenium(IV) dichloride produced the lactone 24 in 80% yield.
M solution of BH3·DMS at −40 °C gave low yields of the reduced product. Whereas, using conc. BH3·DMS solution either in dry toluene or THF furnished the alcohol (21) as a diastereomeric mixture in 6:4 ratio however, the ketone functionality was reduced with Noyori's catalyst Ru[(1S,2S)-p-TsNCH(Ph)CH(Ph)NH](η6-p-cymene) (20)15 in HCOOH affording 21 in 90% yield and 95% de.16 Then, we tried to minimize the use of protecting groups by maintaining the oxygenated functional groups as acetates. Thus, hydrolytic cleavage of acetonide protecting group in 21 leading to a triol, which was acetylated in situ with acetic anhydride in the presence of NEt3 and DMAP to form the tri-acetate 22 in 90% yield for the two steps. The alcohol 22 was transformed into its acrylic ester 23 in 80% yield by treating with acryloyl chloride, catalytic amounts of DMAP and triethylamine in CH2Cl2. Ring closing metathesis17 reaction of 23 in refluxing DCM for 24 h using 10 mol% first generation Grubbs' catalyst, bis(tricyclohexylphosphine)benzylidene ruthenium(IV) dichloride produced the lactone 24 in 80% yield.
We envisioned that partial hydrogenation of the triple bond using Lindlar's catalyst in EtOAc would provide hyptolide. However, this reaction was base as well as time sensitive, maybe it is attributed for the presence of three OAc groups. Hydrogenation using Pd/BaSO4 for 10 min resulted in fully saturated compound by reduction of triple bond and lactone double bond. Use of Pd/CaCO3 in 10 min also resulted in partial hydrogenation of the triple bond to the Z-olefin of along with reduction of the lactone double bond. However, reducing the time to 3 min in the presence of Pd/CaCO3 provided the target lactone, (+)-hyptolide (1) by achieving the partial hydrogenation of the triple bond to the Z-olefin in 90% yield. The synthetic (+)-hyptolide (1) spectroscopic data was identical to that of the natural product.
Overall yields 7.14%, 15.34% and 20.27% reported respectively by Marco & Carda et al.,8a Chakraborty et al.8c and Kumar et al.9 for previous synthetic routes.
Conclusion
We have successfully completed the total synthesis of (+)-hyptolide from 3-butyn-1-ol with an overall yield of 7.08% relying on regioselective epoxide opening, sharpless asymmetric epoxidation, aldehyde alkyne coupling and ring-closing metathesis reactions as key steps.Experimental section
All the solvents and reagents were purified by standard techniques, reactions were performed in oven-dried round bottom flasks; crude products were purified by column chromatography on silica gel (60–120 mesh). Thin layer chromatography plates were visualized by exposure to ultraviolet light and/or by exposure to an methanolic acidic solution of p-anisaldehyde on a hot plate (∼250 °C). Organic solutions were concentrated on rotary evaporator at 40–45 °C. IR spectra were recorded on Perkin–Elmer 683, Thermo Nicolet Nexus 670 spectrometer. 1H NMR and 13C NMR spectra were recorded in CDCl3 solvent on a Varian Gemini 200, Bruker AV-300 and Varian Innova 500 NMR spectrometer. Chemical shifts were reported in parts per million (ppm) with respect to internal TMS. Coupling constants (J) are quoted in Hertz (Hz). s, br s, d, dd, ddd, dt, t, q, qt and m refer to singlet, broad singlet, doublet, doublet of doublet, doublet of doublet of doublet, triplet, quartet, quintet and multiplet, respectively. The optical rotations were measured on a Perkin Elmer-343 polarimeter. The diastereomeric excess of the products were measured by HPLC using Shimadzu LC-20AT series with XDB C18, 150 × 4.6, 5U column. Mass spectra were recorded on Micromass VG-7070H mass spectrometer for ESI and EI are given in mass-to-charge (m/z). High-resolution mass spectra (HRMS) [ESI] were obtained using either a TOF or a double focusing spectrometer.(2S,3R)-5-(Benzyloxy)-1,2-dihydroxypentan-3-yl benzoate (10)
To a stirred solution of epoxy alcohol 9 (7 g, 33.65 mmol) in dry CH2Cl2 (35 mL) containing benzoic acid nuleophile (4.51 g, 37.09 mmol), Ti(O-i-Pr)4 (15.0 mL, 50.52 mmol) was added at room temperature. The mixture was stirred for 1 h at the same temperature. After completion of the reaction, as determined by TLC, the reaction mixture was cooled to 0 °C and quenched with dropwise addition of saturated aqueous NaHCO3 (20 mL). The resulting milky solution was stirred vigorously for 10 h and then filtered through a pad of Celite (CH2Cl2 rinse) and extracted with ethyl acetate (4 × 50 mL). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure and the crude product was purified by silica gel column chromatography (4:6, EtOAc–hexane) to afford 10 (8.32 g, 25.24 mmol, 75%) as a viscous liquid. [α]D25 = +7.4 (c 1.1, CHCl3); IR (KBr): 3420, 2867, 1715, 1275, 1112, 1070, 713 cm−1; 1H NMR (500 MHz, CDCl3):δ 8.01–7.98 (m, 2H), 7.60–7.56 (m, 1H), 7.46–7.41 (m, 2H), 7.37–7.22 (m, 5H), 5.24–5.20 (m, 1H), 4.51 (ABq, J = 17.5, 11.7 Hz, 2H), 3.85–3.80 (m, 1H), 3.74–3.63 (m, 2H), 3.62–3.56 (m, 2H), 2.64 (br s, 1H), 2.27–2.19 (m, 1H), 2.17–2.08 (m, 1H); 13C NMR (CDCl3, 75 MHz): δ 166.4, 137.5, 133.2, 129.6, 128.3, 127.7, 73.2, 72.6, 7 2.2, 66.0, 62.8, 30.9; HRMS (ESI) for C19H22O5Na [M + Na]+ found 353.13594, calcd 353.13531.
(R)-3-(Benzyloxy)-1-((S)-oxiran-2-yl)propyl benzoate (11)
To a stirred solution of 60% sodium hydride dispersion in mineral oil (1.45 g, 60.41 mmol) in THF (17 mL) was added the diol 10 (8.0 g, 24.24 mmol) followed by tosyl-imidazole (10.76 g, 48.46 mmol), and the mixture was stirred for 1 h at 0 °C. After completion of reaction, water was added and extracted with EtOAc (3 × 50 mL). The combined organic fraction was dried over anhydrous Na2SO4, and solvent was removed under reduced pressure. The residue was purified on silica gel column chromatography (3:7 EtOAc–hexane) to afford the epoxide 11 (6.8 g, 21.8 mmol, 90%) as colorless oil. [α]D25 = +14.8 (c 1.0, CHCl3); IR (KBr): 1721, 1269, 1107, 711 cm−1; 1H NMR (500 MHz, CDCl3): δ 8.03–7.99 (m, 2H); 7.59–7.54 (m, 1H); 7.46–7.41 (m, 2H); 7.34–7.20 (m, 5H); 5.21 (td, J = 8.0, 4.8 Hz, 1H), 4.48 (ABq, ΔδAB = 0.02, JAB = 11.9 Hz, 2H), 3.67–3.58 (m, 2H); 3.18–3.15 (m, 1H); 2.83 (dd, J = 5.0 Hz, 1H); 2.79 (dd, J = 5.0 Hz, 1H); 2.18–2.07 (m, 2H); 13C NMR (CDCl3, 75 MHz): δ 165.5, 137.9, 132.9, 129.5, 128.2, 128.1, 127.5, 127.4, 72.9, 70.8, 65.7, 52.3, 45.4, 31.2; HRMS (ESI) for C19H20O4Na [M + Na]+ found 335.12538, calcd 335.12476.
(2S,3R)-5-(Benzyloxy)pentane-2,3-diol (12)
To a stirred suspension of LiAlH4 (2.41 g, 63.42 mmol) in anhydrous THF (15 mL) at 0 °C was added dropwise a solution of terminal epoxide 11 (6.6 g, 21.15 mmol) in anhydrous THF (30 mL). The reaction mixture was allowed to warm to 25 °C and stirred for 3 h. Reaction mixture was then cooled to 0 °C and quenched with dropwise addition of saturated aqueous Na2SO4 (20 mL). The precipitate formed was filtered and washed with ethyl acetate. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure and the crude product was purified by silica gel column chromatography (4:6, EtOAc–hexane) to afford 12 (4.12 g, 19.62 mmol, 93%) as a viscous liquid. [α]D25 = −8.2 (c 0.8, CHCl3); IR (KBr): 3417, 2867, 1367, 1075, 741, 698 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.39–7.28 (m, 5H), 4.54 (s, 2H), 3.83–3.73 (m, 2H), 3.72–3.66 (m, 2H), 3.29 (br s, 1H), 1.90–1.80 (m, 1H), 1.77–1.70 (m, 1H), 1.16 (d, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 125 MHz): δ 137.6, 128.4, 127.7, 127.6, 74.5, 73.2, 69.9, 68.8, 30.5, 17.4; HRMS (ESI) for C12H18O3Na [M + Na]+ found 233.11482, calcd 233.11458.
(4R,5S)-4-(2-(Benzyloxy)ethyl)-2,2,5-trimethyl-1,3-dioxolane (13)
2,2-Dimethoxypropane (3.96 mL, 38.07 mmol) and catalytic PTSA (0.4 g, 2.32 mmol) were added successively to a solution of diol 12 (4 g, 19.04 mmol) in a mixture of CH2Cl2:Me2CO (1:1) (40 mL). The solution was stirred for 10 h at room temperature and then quenched with solid NaHCO3. The crude compound was concentrated in vacuo and purified by column chromatography (1:9, EtOAc–hexane) to afford the acetonide product 13 (4.52 g, 18.08 mmol, 90%) as a colorless liquid. [α]D25 = +30.5 (c 1.0, CHCl3); IR (KBr): 2983, 1374, 1217, 1089, 771, 739 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.35–7.25 (m, 5H), 4.52 (ABq, J = 16.1, 11.9 Hz, 2H), 4.29–4.19 (m, 2H), 3.67–3.58 (m, 2H), 1.82–1.72 (m, 2H), 1.44 (s, 3H), 1.33 (s, 3H), 1.15 (d, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 125 MHz): δ 138.2, 128.0, 127.3, 127.2, 74.7, 73.3, 72.8, 67.1, 30.1, 28.3, 25.6, 15.3; HRMS (ESI) for C15H22O3Na [M + Na]+ found 273.14612, calcd 273.14540.
2-((4R,5S)-2,2,5-Trimethyl-1,3-dioxolan-4-Yl)ethanol (14)
To a stirred solution of naphthalene (13.82 g, 107.96 mmol) in THF (30 mL) were added lithium granules (0.88 g, 125.71 mmol) at room temperature, and the solution was allowed to stir at room temperature for 45 min to generate Li naphthalenide. To the resulting dark green solution was added benzyl ether 13 (4.5 g, 18.0 mmol) at −10 °C, and the mixture was allowed to stir at the same temperature for 1 h, quenched with aqueous NH4Cl, extracted into EtOAc (3 × 50 mL), dried over Na2SO4, concentrated, and purified on silica gel (3:7 EtOAc–hexane) to give alcohol 14 (2.3 g, 14.37 mmol, 81%) as a colorless liquid. [α]D25 = +25.5 (c 0.6, CHCl3); IR (KBr): 3423, 2984, 1376, 1246, 1218, 1085, 1058, 1005, 853 cm−1; 1H NMR (300 MHz, CDCl3): δ 4.35–4.21 (m, 2H), 3.87–3.79 (m, 2H), 2.49 (br s, 1H), 1.85–1.71 (m, 1H), 1.65–1.54 (m, 1H), 1.47 (s, 3H), 1.19 (d, J = 6.0 HZ, 3H); 13C NMR (CDCl3, 75 MHz): δ 107.6, 76.5, 73.5, 60.5, 32.0, 28.2, 25.6, 15.2; HRMS (ESI) for C8H16O3Na [M + Na]+ found 183.0995, calcd 183.0992.
2-((4R,5S)-2,2,5-Trimethyl-1,3-dioxolan-4-yl)acetaldehyde (7)
To a stirred solution of oxalyl chloride (2.33 mL, 27.55 mmol) in dry CH2Cl2 (15 mL) at −78 °C, dry DMSO (3.9 mL, 55.0 mmol) was added dropwise. After 30 min, alcohol 14 (2.2 g, 13.75 mmol) in CH2Cl2 (20 mL) was added over 10 min giving a copious white precipitate. After stirring for 2 h at −78 °C, Et3N (10.97 mL, 82.42 mmol) was added slowly and stirred for 30 min allowing the reaction mixture to warm to rt. The reaction mixture was then diluted with water (20 mL) and expected with CH2Cl2 (3 × 50 mL). The combined organic layer was washed with water (15 mL), brine (10 mL), dried over Na2SO4, and concentrated in vacuo to afford the aldehyde 7, which was directly used for further reaction without purification.(R)-tert-Butyl(5-(4-methoxybenzyloxy)pent-1-yn-3-yloxy)diphenylsilane (16)
Imidazole (3.7 g, 54.41 mmol), and TBDPSCl (7.5 ml, 27.27 mmol) were added to a stirred solution of compound 15 (6.0 g, 27.2 mmol) in CH2Cl2 (35 mL) at 0 °C. Stirring was continued for 10 h and then the mixture was diluted with CH2Cl2 (20 mL). Evaporation of the solvent under reduced pressure followed by column chromatography (1:9, EtOAc–hexane) afforded the silyl ether 16 (11.87 g, 25.91 mmol, 95%) as a colorless liquid. [α]D25 = +11.8 (c 0.7, CHCl3); IR (KBr): 2931, 2857, 1512, 1246, 1108, 702 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.75–7.65 (m, 4H), 7.44–7.33 (m, 6H), 7.15 (d, J = 8.6 HZ, 2H), 6.84 (d, J = 8.6 HZ, 2H), 4.5679 (td, J = 6.4, 1.9 Hz, 1H), 4.30 (s, 2H), 3.80 (s, 3H), 3.63–3.55 (m, 2H), 2.28 (d, J = 2.1 HZ, 1H), 2.07–1.94 (m, 2H), 1.07 (s, 9H); 13C NMR (CDCl3, 125 MHz): δ 159.0, 136.0, 135.8, 134.7, 129.7, 129.6, 129.5, 129.1, 127.6, 127.5, 127.3, 113.6, 84.5, 72.9, 72.4, 65.9, 61.0, 55.2, 38.4, 26.8, 19.2; HRMS (ESI) for C29H34O3NaSi [M + Na]+ found 481.21694, calcd 481.21619.
(R)-3-(tert-Butyldiphenylsilyloxy)pent-4-yn-1-ol (17)
Compound 16 (11.89 g, 25.76 mmol) was taken in 30 mL of CH2Cl2:pH 7 buffer (9:1), DDQ (6.43 g, 28.32 mmol) was added to it, and the solution was stirred for 2 h at room temperature. The reaction mixture was filtered off and the filtrate was washed with 5% NaHCO3 solution (30 mL) and brine (30 mL), dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and purified by silica gel column chromatography (2:8, EtOAc–hexane) to afford 17 as a light yellow colored liquid (7.04 g, 20.82 mmol, 81%). [α]D25 = +33.8 (c 0.6, CHCl3); IR (KBr): 3301, 2957, 2933, 2859, 1108, 1050, 703 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.75 (dt, J = 6.7, 1.3 Hz, 2H), 7.70 (dt, J = 6.5, 1.3 Hz, 2H), 7.44 (dt, J = 7.4, 2.4 Hz, 2H), 7.41–7.37 (m, 4H), 4.59 (td, J = 5.3, 2.1 Hz, 1H), 3.96–3.88 (m, 1H), 3.81–3.74 (m, 1H), 2.36 (d, J = 2.1 Hz, 1H), 2.02–1.96 (m, 1H), 1.89–1.82 (m, 1H), 1.09 (s, 9H); 13C NMR (CDCl3, 125 MHz): δ 135.9, 135.6, 129.8, 129.7, 127.6, 127.3, 83.9, 73.6, 62.0, 59.1, 39.9, 26.7, 19.1; HRMS (ESI) for C21H26O2NaSi [M + Na]+ found 361,15943 calcd 361.15887.
(R)-tert-Butyl(hex-5-en-1-yn-3-yloxy)diphenylsilane (8)
To a stirred solution of oxalyl chloride (3.5 mL, 41.41 mmol) in dry CH2Cl2 (30 mL) at −78 °C, dry DMSO (5.88 mL, 82.81 mmol) was added drop wise. After 30 min, alcohol 17 (7.0 g, 20.71 mmol) in CH2Cl2 (25 mL) was added over 10 min giving a copious white precipitate. After stirring for 2 h at −78 °C, Et3N (16.53 mL, 124.22 mmol) was added slowly and stirred for 30 min allowing the reaction mixture warm to rt. The reaction mixture was then diluted with water (20 mL) and CH2Cl2 (3 × 60 mL). The combined organic layer was washed with water (30 mL), brine (20 mL), dried over Na2SO4, and concentrated in vacuo to afford the aldehyde, which was directly used as such for further reaction.To a stirred suspension of methyltriphenylphosphonium iodide (13.37 g, 33.09 mmol) in dry THF (35 mL) at −78 °C, n-BuLi in hexane (2.0 M, 12.41 mL, 24.82 mmol) was added drop wise under an N2 atmosphere and allowed to return to room temperature. After 45 min, the reaction mixture was again cooled to−78 °C and the aldehyde compound (5.56 g, 16.54 mmol) dissolved in dry THF (20 mL) was added dropwise and stirred for another 45 min. The reaction mixture was quenched with saturated NH4Cl solution (15 mL) at 0 °C and extracted with diethyl ether (2 × 40 mL). The combined organic extracts were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (1:9, EtOAc–hexane) to afford pure compound 8 (3.31 g, 9.91 mmol, 60% over two steps) as a pale yellow liquid. [α]D25 = +16.2 (c 1.0, CHCl3); IR (KBr): 2933, 2858, 1109, 1082, 702 cm−1; 1H NMR (300 MHz, CDCl3): δ 7.77–7.66 (m, 4H), 7.48–7.32 (m, 6H), 5.91–5.76 (m, 1H), 5.10–5.00 (m, 2H), 4.37 (td, J = 5.2, 2.2 Hz, 1H), 2.50–2.35 (m, 2H), 2.34 (d, J = 1.5, Hz, 1H), 1.08 (s, 9H); 13C NMR (CDCl3, 125 MHz): δ 135.9, 135.8, 133.2, 129.7, 129.6, 127.5, 127.4, 117.9, 84.4, 73.0, 63.3, 42.7, 26.8, 19.2; anal. calcd for C22H26OSi: C, 78.99; H, 7.83%; found: C, 78.60; H, 7.77%.
(R)-5-(tert-Butyldiphenylsilyloxy)-1-((4R,5S)-2,2,5-trimethyl-1,3-dioxolan-4-yl)oct-7-en-3-yn-2-one (19)
To a stirred solution of alkyne 8 (2 g, 5.98 mmol) in dry THF (20 mL) was slowly added n-BuLi (2.0 M, 4.5 mL, 8.98 mmol, solution in hexanes) at −20 °C under N2. The reaction mixture was stirred for 30 min at −78 °C, and a solution of compound 7 (1.13 g, 7.15 mmol) in dry THF (15 mL) was added dropwise with stirring. The mixture was kept at −40°C for 2 h and then allowed to warm to rt for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution, and extracted with EtOAc, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (hexane–EtOAc = 75:25) to afforded alcohol 18 as a 1:1 mixture of diastereomers (determined by chiral HPLC analysis) in approximately 65% yield.
To the above obtained alcohol 18 (1.8 g, 3.65 mmol) in 20 mL of dry CH2Cl2, Dess–Martin periodinate (2.32 g, 5.46 mmol) and NaHCO3 (0.92 g, 10.95 mmol) were added at 0 °C and stirred for 2 h. After completion of the reaction, the reaction was quenched with aqueous sodium thiosulfate solution (6 mL) and saturated aqueous sodium bicarbonate solution (10 mL). The reaction mixture was extracted with CH2Cl2 (2 × 20 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (3:7, EtOAc–hexane) to afford 19 (1.46 g, 2.97 mmol, 82%) as a yellow liquid. [α]D25 = +50.1 (c 0.5, CHCl3); IR (KBr): 2983, 2924, 2860, 1679, 1109, 1083, 704 cm−1; 1H NMR (300 MHz, CDCl3): δ 7.76–7.65 (m, 4H), 7.46–7.35 (m, 6H), 5.91–5.74 (m, 1H), 5.16–5.05 (m, 2H), 4.56–4.44 (m, 2H), 4.29 (t, J = 6.0 Hz, 1H), 2.66–2.35 (m, 4H), 1.41 (s, 3H), 1.32 (s, 3H), 1.12–1.03 (m, 12H); 13C NMR (CDCl3, 125 MHz): δ 184.2, 135.8, 135.7, 134.4, 130.0, 129.8, 129.5, 118.6, 107.7, 92.8, 83.7, 73.4, 73.0, 63.3, 46.1, 41.9, 28.2, 26.7, 25.6, 19.2, 15.2; HRMS (ESI) for C30H38O4NaSi [M + Na]+ found 513.24316, calcd 513.24268.
(2S,5R)-5-(tert-Butyldiphenylsilyloxy)-1-((4R,5S)-2,2,5-trimethyl-1,3-dioxolan-4-yl)oct-7-en-3-yn-2-ol (21)
To a mixture of propargyl ketone 19 (1.0 g, 2.04 mmol) in formic acid (0.77 mL, 20.21 mmol) and triethylamine (1.13 mL, 8.15 mmol) were added at room temperature an aliquant amount of the stock solution of the RuCl[N-(tosyl)-(1,2-diphenylethylenediamine)(p-cymene)] complex 0.0337 M in CH2Cl2 (1.51 mL, 0.051 mmol), prepared according to ref. 15. The reaction mixture was stirred at room temperature for overnight. The reaction was quenched with saturated aqueous NaHCO3 solution, and extracted with CH2Cl2, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (3:7, EtOAc–hexane) to afford chiral propargyl alcohol 21 (0.9 g, 1.82 mmol, 90%) as a light yellow colored liquid. [α]D25 = +54.6 (c 0.9, CHCl3); IR (KBr): 3448, 2933, 2892, 1109, 1081, 705 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.77–7.73 (m, 2H), 7.72–7.68 (m, 2H), 7.45–7.34 (m, 6H), 5.91–5.81 (m, 1H), 5.11–5.04 (m, 2H), 4.47 (td, J = 6.1, 1.3 Hz, 1H), 4.42–4.37 (m, 1H), 4.25–4.18 (m, 1H), 4.10–4.04 (m, 1H), 2.52–2.38 (m, 2H), 1.83–1.67 (m, 2H), 1.43 (s, 3H), 1.31 (s, 3H), 1.12 (d, J = 6.4 Hz, 3H), 1.06 (s, 9H); 13C NMR (CDCl3, 125 MHz): δ 135.9, 135.7, 133.5, 129.7, 129.5, 127.5, 127.2, 117.7, 107.8, 85.4, 73.4, 76.1, 73.4, 63.5, 61.1, 42.7, 37.6, 28.3, 26.7, 25.6, 19.2, 15.2; HRMS (ESI) for C30H40O4NaSi [M + Na]+ found 515.25881, calcd 515.25707.
(2S,3R,5S,8R)-8-(tert-Butyldiphenylsilyloxy) undec-10-en-6-yne-2,3,5-triyl triacetate (22)
To a stirred solution of compound 21 (0.8 g, 1.62 mmol) in a MeOH (20 mL) and was added PTSA (0.2 g, 1.16 mmol) under N2, then the mixture was stirred at room temperature for 8 h. The mixture was quenched with solid NaHCO3 (0.2 g) and filtered, the solvent was removed under reduced pressure and the crude triol was directly used for the next step without further purification.Anhydrous Et3N (0.92 mL, 6.63 mmol), Ac2O (0.33 mL, 3.23 mmol), and DMAP (10 mg, 0.082 mmol) were added to a solution of triol (0.5 g, 1.10 mmol) in anhydrous CH2Cl2 (15 mL) under nitrogen atmosphere at room temperature. The mixture was stirred at room temperature for 45 min. The solvent was removed under reduced pressure, and the mixture was purified by silica gel column chromatography (2:8, EtOAc–hexane) to afford 22 (0.57 g, 0.98 mmol, 90% over two steps) as a light yellow colored liquid.[α]D25 = +16.0 (c 0.9, CHCl3); IR (KBr): 1745, 1370, 1228, 1109, 1079, 1023, 702 cm; 1H NMR (500 MHz, CDCl3): δ 7.72 (dd, J = 7.9 Hz, 2H), 7.67 (dd, J = 7.9 Hz, 2H), 7.44–7.35 (m, 6H), 5.86–5.76 (m, 1H), 5.31 (td, J = 5.9, 1.3 Hz, 1H), 5.09–5.02 (m, 4H), 4.39 (td, J = 6.5, 1.2 Hz, 1H), 2.47–2.35 (m, 2H), 2.03 (s, 3H), 2.02 (s, 3H), 2.0 (s, 3H), 1.28–1.24 (m, 2H), 1.17 (d, J = 6.4 Hz, 3H), 1.06 (s, 9H); 13C NMR (CDCl3, 75 MHz): δ 170.1, 169.9, 169.4, 135.9, 135.7, 133.2, 129.7, 129.5, 127.5, 127.3, 117.9, 87.0, 81.1, 71.4, 70.0, 63.3, 61.2, 42.5, 33.9, 26.7, 21.0, 20.8, 19.2, 15.2; HRMS (ESI) for C33H42O7NaSi [M + Na]+ found 601.25920, calcd 601.25842.
(2S,3R,5S,8R)-8-Hydroxyundec-10-en-6-yne-2,3,5-triyl triacetate (6)
A 1 M solution of TBAF in THF (1.73 mL, 1.73 mmol) was added to a solution of compound 22 (0.5 g, 0.86 mmol) in dry THF (15 mL) at 0 °C. The mixture was stirred at room temperature for 3 h. After completion of the reaction, the mixture was diluted with EtOAc (15 mL). The combined organic layers were washed with sat. NaCl, and the mixture was extracted with ethyl acetate (3 × 10 mL). The organic extracts were washed with brine and dried over Na2SO4. The solvent was removed under reduced pressure, and the mixture was purified by silica gel column chromatography (3:7, EtOAc–hexane) to afford 6 (0.2 g, 0.58 mmol, 70%) as a colorless liquid. [α]D25 = −21.5 (c 0.7, CHCl3); IR (KBr): 3465, 1741, 1372, 1230, 1024 cm−1; 1H NMR (300 MHz, CDCl3): δ 5.94–5.78 (m, 1H), 5.45 (dq, J = 5.4, 1.5 Hz, 1H), 5.31–5.03 (m, 4H), 4.43 (t, J = 5.4 Hz, 1H), 2.62 (brs, 1H), 2.50–2.42 (m, 2H), 2.19–2.10 (m, 1H), 2.09 (s, 3H), 2.08 (s, 3H), 2.04 (s, 3H), 2.02–1.92 (m, 1H), 1.22 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 75 MHz): δ 170.4, 170.1, 169.5, 132.7, 118.8, 87.4, 80.8, 71.2, 70.4, 61.3, 61.2, 41.7, 34.1, 21.0, 20.9, 20.8, 14.7; HRMS (ESI) for C17H24O7Na [M + Na]+ found 363.14142 calcd 363.14077.
(2S,3R,5S,8R)-8-(Acryloyloxy)undec-10-en-6-yne-2,3,5-triyl triacetate (23)
Acryloyl chloride (0.058 mL, 0.74 mmol) was added drop wise under N2 to a solution of alcohol 6 (0.17 g, 0.5 mmol), Et3N (0.13 mL, 1.0 mmol), and DMAP (5 mg, 0.041 mmol) in anhyd CH2Cl2 (10 mL). The mixture was stirred at room temperature for 1 h until the reaction was complete (TLC). The mixture was then poured into brine and extracted with CH2Cl2 (2 × 10 mL). The organic phases were washed with 1 M aq. HCl (5 mL) and brine (8 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude product was purified by column chromatography (2:8, EtOAc–hexane) to give 23 (0.15 g, 0.38 mmol, 80%) as a light yellow liquid. [α]D25 = +10.8 (c 0.7, CHCl3); IR (KBr): 2984, 2930, 1733, 1228, 1181, 1083, 1024 cm−1; 1H NMR (300 MHz, CDCl3): δ 6.47 (dd, J = 30.6, 17.2 Hz, 1H), 6.18–6.08 (m, 1H), 5.90–5.75 (m, 2H), 5.53–5.43 (m, 2H), 5.19–5.06 (m, 4H), 2.56 (t, J = 6.5 Hz, 2H), 2.19–2.09 (m, 1H), 2.08 (s, 3H), 2.06 (s, 3H), 2.04 (s, 3H), 2.03–1.96 (m, 1H), 1.20 (d, J = 6.5 Hz, 3H); 13C NMR (CDCl3,75 MHz): δ 170.2, 170.1, 169.5, 164.8, 131.6, 127.8, 118.9, 83.3, 81.9, 71.3, 70.1, 63.0, 61.1, 38.8, 33.9, 21.0, 20.9, 15.2; HRMS (ESI) for C20H26O8Na [M + Na]+ found 417.15199 calcd 417.15074.
(2S,3R,5S)-7-((R)-6-oxo-3,6-Dihydro-2H-pyran-2-yl)hept-6-yne-2,3,5-triyl triacetate (24)
Grubbs' first-generation catalyst (0.033 g, 0.0415 mmol) was added to a solution of 23 (0.1 g, 0.25 mmol) in anhyd. CH2Cl2 (25 mL) at 0 °C, and the mixture was allowed to warm to room temperature over 24h. After completion of the reaction (TLC), the solvent was removed under reduced pressure and the residue was purified by column chromatography (5:5, EtOAc–hexane) to give 24 (0.073 g, 0.19 mmol, 80%) as a colorless liquid. [α]D25 = −4.0 (c 0.3, CHCl3); IR (KBr): 2924, 2853, 1739, 1374, 1231, 1049, 1025 cm−1; 1H NMR (500 MHz, CDCl3): δ 6.90 (dt, J = 9.7, 4.2 Hz, 1H), 6.08 (dt, J = 9.9, 1.8 Hz, 1H), 5.40 (dddd, J = 14.8, 9.1, 5.4, 1.3 Hz, 1H), 5.21 (td, J = 7.0, 1.2 Hz, 1H), 5.17 (dt, J = 10.2, 3.0 Hz, 1H), 5.08–5.03 (m, 1H), 2.70–2.65 (m, 2H), 2.17–2.10 (m, 1H), 2.09 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H), 2.03–1.96 (m, 1H), 1.2 (d, J = 6.5 Hz, 3H); 13C NMR (CDCl3, 75 MHz): δ 170.2, 169.5, 162.2, 144.1, 121.3, 82.7, 82.4, 71.1, 70.2, 66.7, 60.9, 33.8, 30.9, 21.0, 20.9, 20.8, 15.1; HRMS (ESI) for C18H22O8Na [M + Na]+ found 398.12069 calcd 389.12062.
6R-(1Z,3S,5R,6S)-5,6-Dihydro-6-[3,5,6-tris(acetoxy)-1-heptenyl]-2H-pyran-2-one (hyptolide) (1)
To solution of compound 24 (0.04 g, 0.10 mmol) in EtOAc (10 mL), one drop of quinoline and Lindlar's catalyst (Pd/CaCO3) were added and stirred at room temperature under H2 atm for 3min. After completion of the reaction, the reaction mixture was filtered and the solvent was removed under reduced pressure. The crude product was purified on silica gel column chromatography (ethyl acetate–hexane, 5:5) to give hyptolide 1 (32 mg, 0.087 mmol, 80%) as a solid, mp 83–87 °C, (ref. 3 m.p. 87–88 °C); [α]D25 = +12.5 (c 0.7, CHCl3); {ref. 3 [α]D25 = +11.2 (c = 0.6, CHCl3)}. IR (KBr): 1738, 1373, 1238, 1045 cm−1; 1H NMR (500MHz, CDCl3): δ 6.91–6.86 (m, 1H), 6.07–6.03 (m, 1H), 5.81–5.76 (m, 1H), 5.57–5.50 (m, 2H), 5.32–5.25 (m, 1H), 5.02–4.96 (m, 1H), 4.92 (dt, J = 9.4, 3.2 Hz, 1H), 2.48–2.38 (m, 2H), 2.10–2.06 (m, 4H), 2.05 (s, 3H), 2.03 (s, 3H), 1.87–1.80 (m, 1H), 1.20 (d, J = 6.5 Hz, 3H). 13C NMR (CDCl3, 75MHz): δ 170.5, 170.2, 165.0, 163.3, 144.5, 131.2, 130.7, 121.5, 73.8, 70.9, 70.4, 66.5, 34.8, 30.9, 21.2, 21.1, 21.0, 14.7; HRMS (ESI) for C18H24O8Na [M + Na]+ found 391.13634 calcd 391.13530.
Acknowledgements
AR and CNR thank UGC, New Delhi for the award of fellowships. The authors thank CSIR, New Delhi for financial support as part of XII Five Year plan programme under title ORIGIN (CSC-0108).Notes and references
- K. Gorter, Bull. Jard. Bot. Buitenzorg, 1920, 1, 327 Search PubMed.
- A. J. Birch and D. N. Butler, J. Chem. Soc., 1964, 4167–4168 RSC.
- S. A. Achmad, T. Høyer, A. Kjær, L. Makmur and R. Norrestam, Acta Chem. Scand., 1987, 41B, 599–609 CrossRef.
- R. Pereda-Miranda, M. Fragoso-Serrano and C. M. Cerda-García-Rojas, Tetrahedron, 2001, 57, 47–53 CrossRef CAS.
- M. T. Davies-Coleman, R. B. English and D. E. A. Rivett, Phytochemistry, 1987, 26, 1497–1499 CrossRef.
- L. A. Collett, M. T. Davies-Coleman and D. E. A. Rivett, Phytochemistry, 1998, 48, 651–656 CrossRef CAS.
- A. Alemany, C. Márquez, C. Pascual, S. Valverde, M. Martínez-Ripoll, J. Fayos and A. Perales, Tetrahedron Lett., 1979, 20, 3583–3586 CrossRef.
- (a) J. Murga, J. García-Fortanet, M. Carda and J. A. Marco, Tetrahedron Lett., 2003, 44, 1737–1739 CrossRef CAS; (b) J. García-Fortanet, J. Murga, M. Cardaa and J. A. Marco, Tetrahedron, 2004, 60, 12261–12267 CrossRef; (c) T. K. Chakraborty and S. Purkait, Tetrahedron Lett., 2008, 49, 5502–5504 CrossRef CAS.
- P. S. Chowdhury and P. Kumar, Eur. J. Org. Chem., 2013, 4586–4593 CrossRef CAS.
- For our recent contributions to δ-lactone-containing natural products: (a) G. Sabitha, K. Shankaraiah and J. S. Yadav, Eur. J. Org. Chem., 2013, 4870–4878 CrossRef CAS; (b) G. Sabitha, S. K. Das, A. Praveen and J. S. Yadav, Tetrahedron Lett., 2013, 54, 1097–1099 CrossRef CAS; (c) G. Sabitha, D. V. Reddy, S. S. S. Reddy, J. S. Yadav, C. G. Kumar and P. Sujitha, RSC Adv., 2012, 2, 7241–7247 RSC; (d) G. Sabitha, A. S. Rao and J. S. Yadav, Tetrahedron: Asymmetry, 2011, 22, 866–871 CrossRef CAS; (e) G. Sabitha, S. S. S. Reddy and J. S. Yadav, Tetrahedron Lett., 2011, 52, 2407–2409 CrossRef CAS; (f) G. Sabitha, S. S. S. Reddy, A. Raju and J. S. Yadav, Synthesis, 2011, 1279–1282 CrossRef CAS; (g) G. Sabitha, C. N. Reddy, A. Raju and J. S. Yadav, Tetrahedron: Asymmetry, 2011, 22, 493–498 CrossRef CAS; (h) G. Sabitha, C. N. Reddy, P. Gopal and J. S. Yadav, Tetrahedron Lett., 2010, 51, 5736–5739 CrossRef CAS; (i) G. Sabitha, M. Bhikshapathi, N. Ranjith, N. Ashwini and J. S. Yadav, Synthesis, 2011, 821–825 CrossRef CAS.
- J. S. Yadav, M. Raj Kumar and G. Sabitha, Tetrahedron Lett., 2008, 49, 463–466 CrossRef CAS.
- C. Maurice and K. B. Sharpless, J. Org. Chem., 1985, 50, 1557–1560 CrossRef.
- R. D. Cink and C. J. Forsyth, J. Org. Chem., 1995, 60, 8122–8123 CrossRef CAS.
- (a) G. Sabitha, K. Sudhakar and J. S. Yadav, Tetrahedron Lett., 2006, 47, 8599–8602 CrossRef CAS; (b) G. Sabitha, V. Bhaskar, S. S. S. Reddy and J. S. Yadav, Tetrahedron, 2008, 64, 10207–10213 CrossRef CAS; (c) G. Sabitha, V. Bhaskar, S. S. S. Reddy and J. S. Yadav, Chin. J. Chem., 2010, 28, 2421–2424 CrossRef CAS.
- (a) J. A. Marshall and K. Ellis, Tetrahedron Lett., 2004, 45, 1351–1353 CrossRef CAS; (b) M. Li and G. A. O.-Doherty, Tetrahedron Lett., 2004, 45, 6407–6411 CrossRef CAS.
- The diastereomeric excess of the product was determined using a Shimadzu high-performance liquid-chromatography (HPLC) system equipped with a chiral HPLC column (XDB C18, 150 × 4.6, 5U) and a UV detector at an absorbance of 225 nm. ZORBAX SBC 18 250 × 4.6 mm, 5μ (column) and a solvent system of acetonitrile and water (3:1) at a flow rate of 1.0 mL per min were used. tR: 21.17 and 24.34 min.
- For a recent review on ring-closing metathesis reactions, see: (a) A. Fürstner, Angew. Chem., Int. Ed., 2000, 39, 3012–3043 CrossRef; (b) T. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS PubMed; (c) S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900–1923 CrossRef CAS PubMed; (d) A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243–251 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all compounds. See DOI: 10.1039/c3ra45042b |
| This journal is © The Royal Society of Chemistry 2014 |