
Oxygen defects and formation of Ce3+ affecting the photocatalytic performance of CeO2 nanoparticles†
Abstract
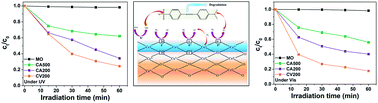
Here we report the photocatalytic activity of CeO2 nanoparticles. This is carried out with methyl orange as the reference pollutant. Annealing of ceria under vacuum generates oxygen deficient CeO2 nanoparticles with defects such as oxygen vacancies and formation of Ce3+. This is evident from the characterization results of X-ray diffraction, Raman spectroscopy, N2 adsorption–desorption and X-ray photoelectron spectroscopy. The band gap is red shifted due to the creation of intermediate energy states of Ce3+ and oxygen vacancies in the band gap. The reduced photoluminescence (PL) intensity of defective ceria indicates that the electron–hole separation is substantially enhanced by the surface trap centers. Air annealed ceria not only has relatively low surface area but also has fewer surface defects. Thus, it is expected to display less photocatalytic activity. Vacuum annealed CeO2 indeed displays better photocatalytic activity in the degradation of methyl orange under UV and visible light as compared to the air annealed samples.
 Please wait while we load your content...
Please wait while we load your content...

