 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A microfluidic system supports single mouse embryo culture leading to full-term development†
Telma Cristina
Esteves
a,
Fleur
van Rossem
b,
Verena
Nordhoff
c,
Stefan
Schlatt
c,
Michele
Boiani
a and
Séverine
Le Gac
*b
aMax-Planck Institute for Molecular Biomedicine, Röntgenstrasse 20, 48149 Münster, Germany
bBIOS, Lab on a Chip group, MESA+ Institute for Nanotechnology, University of Twente, P.O. box 217, 7500AE Enschede, The Netherlands. E-mail: s.legac@utwente.nl; Fax: +31 534893595; Tel: +31 534892722
cCentre of Reproductive Medicine and Andrology, University Hospital of Münster, Domagkstrasse 11, 48149 Münster, Germany
First published on 21st October 2013
Abstract
The present study demonstrates the feasibility of application of a microfluidic system for in vitro culture of pre-implantation mouse embryos, with subsequent development to full-term upon embryo transfer. Specifically, embryos cultured in groups in nL volume chambers achieve pre-implantation developmental rates up to 95% (4.5 days after fertilization), while birth rates upon transfer in utero are comparable to conventional droplet culture (∼30%). Importantly, while culturing single embryos in conventional microliter droplets hampers full-term development, mouse embryos cultured individually in a confined microfluidic environment achieve normal birth rates (29–33%) with normal morphology. Furthermore, the refreshment of culture media (dynamic culture) during pre-implantation in the microfluidic system does not impair development to term. These results deliver great promise to studies in developmental biology and human assisted reproductive technologies (ART), as nanoliter culture volumes provided by microfluidics will (1) allow online screening of physical and chemical culture parameters and (2) facilitate the acquisition of physiological data at the single embryo level – essential requisites for the determination of optimal embryo culture conditions.
Introduction
The field of assisted reproductive technologies (ART) is a fast growing area, currently facing the double challenge of satisfying an increasing demand while improving the success rate of treatments. In this context, clinics encounter problems transcending the inherent sub-fertility condition of the treated couples. Notably, the protocol employed for the in vitro culture of pre-implantation embryos is considered sub-optimal.1,2 This culture protocol has been adapted from the routines developed for in vitro experimentation on somatic cells, as the natural embryo environment and its physiological needs are extremely hard to assess.1 Furthermore, culture formats such as 5–20 μL droplets,1,2 glass oviducts,3 and well-of-the-well devices,4,5 designed to provide environments physically close to the natural one – the oviduct – have not so far helped achieve better results.2 Consequently, fundamental research is urgently needed to get a better insight into the impact of culture conditions on the embryo viability, and to identify optimal physical and chemical parameters for the in vitro culture of pre-implantation embryos.Here, we propose that microfluidics can contribute to this decisive issue of the field of ART by offering a customized microenvironment that resembles that found in the oviduct, as well as integrated platforms with parallelization potential for single embryo manipulation with direct feedback on their development. First, the micrometer-sized dimensions found in microdevices lend themselves well to fine-tuning of the physical and chemical microenvironment of the culture,6 to the creation of a dynamic environment as well as spatial and temporal gradients,7 as found in vivo. Also, this format is ideal for cell experimentation down to the single cell level,8 and has proven its suitability for studies on embryos of various species, down to the single embryo level.9–11 Finally, microfluidic constructs can easily be multiplexed and include miniaturized sensors to monitor in situ specific physical or (bio)chemical parameters. So far, despite its tremendous potential, microfluidics has not yet been appropriately explored for embryo culture: embryos have only been cultured in groups in microfluidic devices,12–14 in unconfined volumes of medium,15–17 and attempts to realize (semi)-dynamic culture12,18 has resulted in embryo viability loss.
Here, we present the first-ever study in which microfluidic and droplet culture system are compared side by side for their ability to support the entire development of mouse embryos, using embryo densities down to one (as is the case in human ART). Various culture conditions are screened and their impact on the embryo development is assessed, including full-term development (Fig. 1). Specifically, mouse embryos are cultured throughout their pre-implantation development, from the zygote stage until 3.5–4.5 days post coitum (dpc) in both nL chambers (30 or 270 nL) and conventional drops, individually or as groups (5 or 20). In one set of experiments (groups of 5 embryos, microfluidics), medium is replenished at 3 dpc, as to determine whether medium exchange affects embryo development. Embryo developmental competence is assessed in terms of pre-implantation development (at 3.5 and 4.5 dpc) and cell count (at 4.5 dpc), as well as full-term development after embryo transfer in pseudo-pregnant recipient mice. The proposed microfluidic approach combined with single embryo culture opens novel possibilities for the field of developmental and reproductive biology, towards the identification of optimal in vitro culture conditions and the elucidation of the embryo's physiology.
 | ||
| Fig. 1 Experimental design. Naturally fertilized mouse oocytes (B6C3F1xCD1) are collected from copulated mice after mating (18 h post-hCG), randomly allocated to various group cultures, and cultured in vitro from the zygote stage till the blastocyst stage (3.5–4.5 dpc) in α-MEM medium. Culture groups are characterized by the embryo number (1, 5 or 20), the culture format employed for their in vitro development (microfluidic system or droplet), the volume employed for the culture (30 nL, 270 nL or 5 μL), and by the implementation or not of a medium refreshment step at day 3 after fertilization. At 3.5 dpc, a subset of embryos is retrieved from the microfluidic chambers and droplets and transferred to the uterus of pseudo-pregnant mice to assess their full-term competence. Embryo growth is assessed at various time-points: during the pre-implantation period at 3.5 dpc and 4.5 dpc, for their developmental stage; at 4.5 dpc, for the number of cells; and for their full-term development. Allocation of the embryos to the different experimental groups is detailed in Tables 1 and 2. | ||
Materials and methods
Microsystem fabrication
Microfluidic devices are fabricated in PDMS (polydimethylsiloxane) using standard soft lithography techniques.19 Briefly, a silicon-based mould is first fabricated in the cleanrooms of the MESA + Institute for Nanotechnology using photolithography and dry-etching techniques, based on a design drawn with Clewin (WieWeb software, Hengelo, The Netherlands). This mould is subsequently coated with FDTS (perfluorodecyltrichlorosilane; ABCR, GmBH, Germany) to decrease the surface energy and facilitate the removal of the PDMS layer. Thereafter, a 10:1 PDMS pre-polymer:curing agent mixture (Sylgard 184, Dow Corning GmbH, Wiesbaden, Germany) is prepared and thoroughly degassed, poured on the mould, degassed once more and let for curing overnight at 60 °C. Following this, the PDMS layer is released from the mould, cut into individual devices using a sharp knife, and reservoirs are punched with sharp needles. Finally, the devices are bonded to glass cover slips using oxygen plasma treatment.Flow characterization and medium refreshment protocol
The technique of passive pumping20 is employed for fluid manipulation in the device and medium refreshment. The flow profile and flow-rates are determined experimentally using 3 μm diameter Polybead Polystyrene Violet Dyed beads (Polysciences Inc, Warrington, Pennsylvania, USA). Based on the passive pumping technique, a protocol for complete medium refreshment in the device is developed using fluorescent dyes (fluorescein sodium salt, and rhodamine B; both purchased from Sigma, Zwijndrecht, The Netherlands). The device is initially filled in with a fluorescein solution and droplets (1.5 μL) of a rhodamine B solution are applied in the inlet reservoir and pumped in the device until no green fluorescence is detected anymore in the nL chamber. These characterization experiments are carried out in empty chambers and chambers loaded with 80 μm microbeads (Megabead NIST Traceable Particle Size Standard, Polysciences Inc, Warrington, Pennsylvania USA), which are used as surrogates for mouse embryos.Animal handling
Mice are maintained and handled at the animal facility of the Max-Planck-Institute in Münster, according to the breeding/keeping licence and the ethical permit issued by the Landesamt für Natur, Umwelt und Verbraucherschutz (LANUV) of the state of North Rhine-Westphalia, Germany. Mice are housed in groups of five, exposed to 14L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10D (light:dark) hours photoperiod, and are fed ad libitum on Harlan-Teklad 2020SX diet (Harlan Laboratories, Oxford, United Kingdom). Five to seven week old females of the B6C3F1 strain (C57Bl/6J x C3H/HeN) are superovulated by intra-peritoneal injection of 10 international units (IU) of pregnant mare's serum gonadotropin (PMSG; Intergonan, Intervet, Germany), followed by 10 IU of human chorionic gonadotropin (hCG; Ovogest, Intervet, Germany) 48 h later. Immediately after hCG injection, females are mated to CD1 stud males. Copulated females (identified the next morning by presence of a vaginal plug) are sacrificed after cervical dislocation 17 h after hCG injection, and 1-cell embryos (zygotes) are retrieved from the oviductal ampullae. Zygotes are handled in Hepes-buffered CZB (HCZB) medium, as previously described,21,22 and are separated from any remaining cumulus cells by treatment with 50 IU mL−1 hyaluronidase (ICN Biomedicals, Eschwege, Germany) in HCZB medium, followed by gentle pipetting.
:10D (light:dark) hours photoperiod, and are fed ad libitum on Harlan-Teklad 2020SX diet (Harlan Laboratories, Oxford, United Kingdom). Five to seven week old females of the B6C3F1 strain (C57Bl/6J x C3H/HeN) are superovulated by intra-peritoneal injection of 10 international units (IU) of pregnant mare's serum gonadotropin (PMSG; Intergonan, Intervet, Germany), followed by 10 IU of human chorionic gonadotropin (hCG; Ovogest, Intervet, Germany) 48 h later. Immediately after hCG injection, females are mated to CD1 stud males. Copulated females (identified the next morning by presence of a vaginal plug) are sacrificed after cervical dislocation 17 h after hCG injection, and 1-cell embryos (zygotes) are retrieved from the oviductal ampullae. Zygotes are handled in Hepes-buffered CZB (HCZB) medium, as previously described,21,22 and are separated from any remaining cumulus cells by treatment with 50 IU mL−1 hyaluronidase (ICN Biomedicals, Eschwege, Germany) in HCZB medium, followed by gentle pipetting.
Culture of mouse embryos in microfluidic chambers and droplets
Mouse embryo culture experiments are conducted over several days and using different batches of microfluidic systems (different experimental series), in order to assess biological and microdevice reproducibility. Microfluidic systems are washed with 70% ethanol, twice with fresh Milli Q water prior to use, and placed in 35 mm Petri-dishes (Sarstedt, Nümbrecht, Germany). 2 mL of alpha-modified Eagle's medium23,24 (α-MEM, M4526 Sigma, Germany) supplemented with 0.2% w/v BSA (Pentex; Serological Proteins Inc., Kankakee, IL, USA) and 50 μg mL−1 gentamicin sulphate (ICN Biomedicals, Irvine, CA, USA) are finally added in the Petri dish around the microsystem to preserve a high level of humidity and prevent medium evaporation in the device. The reduced final medium volume is thereby kept below reservoir level, avoiding contact with the surface and interior of the microfluidic system. Devices are filled with the same culture medium and pre-incubated at 37 °C under 7% CO2. Zygotes are placed in the inlet reservoir using a curved polished glass pipette operated through a mouthpiece, and subsequently transferred to the culture chamber using passive pumping.20 Embryos are cultured at 37 °C under 7% CO2 until further manipulation. For one set of experiments, medium is replenished in the microfluidic chambers (“dynamic culture”), as described in the previous section. For that purpose, medium is maintained at culture conditions (37 °C under 7% CO2) prior to medium exchange. When outside the incubator, microfluidic systems are operated in a room at 30 °C as quickly as possible. Thereby, in terms of physical conditions, dynamic culture experiments follow the protocols used in conventional embryo culture as closely as possible. For control experiments, conditions used usually in embryology studies as well as ART culture are used: embryos are grown in 5 μL droplets (in the same culture medium), placed in a sterile Petri-dish and covered with mineral oil suitable for mouse embryo culture (Sigma M8410), to prevent medium evaporation. Embryo development during culture in both microfluidic chambers and droplets is imaged using a stereomicroscope (Nikon SMZ800, Plan Apo 1X WD70, Düsseldorf, Germany) coupled to a camera (Nikon, DS-2Mv) and a computerized image acquisition system (ACT-2U Version 1.51.116.256).Embryo transfer
Embryos are retrieved from microfluidic systems at 3.5 dpc by applying 10 μL of culture medium on the outlet reservoir to create a mild back-flow in the chamber. Thereby, the embryos are displaced through the V-shape structures into the inlet reservoir. There, embryos are collected using a flame-polished glass pipette and placed in 400 μL of HCZB medium (Nunc 4-well dish; Thermo Fisher Scientific, Langenselbold, Germany), until further processing. Embryos are selected for embryo transfer (ET) on the basis of the presence of a cavity (blastocoel), which is indicative of the blastocyst stage; in a situation where embryos would progress in pre-implantation development at a slower rate (i.e., reaching at least the morula stage, not showing a cavity), morula embryos (no cavity) have also been used for ET (random sampling). Embryo transfer is performed as previously described.21,22 Blastocysts are transferred in groups of 9–11 embryos to the left uterine horn of each pseudo-pregnant female (one experimental condition per female). Pregnancies are allowed to progress to term.Blastocyst cell count
After retrieval, blastocysts are stained at 4.5 dpc for cell count using nucleus staining (DRAQ5). Imaging is performed with an UltraVIEW RS 3ES spinning disk confocal imaging system (PerkinElmer LAS GmbH, Jügesheim, Germany) on an inverted microscope (TE-2000U, Nikon GmbH, Düsseldorf, Germany) using a 60x water immersion lens (N.A. 1.2). Acquired images are imported into Volocity (PerkinElmer, Waltham, MA, USA) and processed for cell counting (based on nuclear staining) using maximum intensity projections.Statistical analysis
Blastocyst and birth rates (i.e., proportions) are analyzed according to Norman TJ Bailey (Statistical methods in biology, 1995, Cambridge University Press). Briefly, given two groups, one of n1 embryos with a1 having a certain character (e.g., blastocyst formation), the other of n2 embryos with a2 having the character, the mathematical formula of Bailey answers the question of whether the two groups may be considered as drawn from the same population. First, the following parameters are calculated: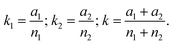
Subsequently, d the units of standard deviation that separate the two groups from each other is determined: 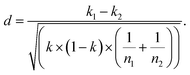 d is referred to the areas of the normal curve. Values of d higher than 1.960 will cause to reject the null hypothesis at a confidence level of 5% (two tails).
d is referred to the areas of the normal curve. Values of d higher than 1.960 will cause to reject the null hypothesis at a confidence level of 5% (two tails).
Results
Microfluidic environment for embryo pre-implantation culture
To more closely recapitulate the physical confinement found in the mouse oviduct, a microfluidic system is developed that includes a culture chamber with nanoliter capacity (30 or 270 nL; see Fig. 2). Two reservoirs are connected to the chamber via microchannels for the introduction of solutions and embryos. Importantly, this configuration allows diffusion-based delivery of nutrients from the reservoirs of the device as well as accumulation of factors secreted by the embryos.25 Altogether, gradients are established during the culture in the vicinity of the embryo(s), mimicking thereby conditions found in vivo. Furthermore, structures are added at the inlet and outlet of the chamber to trap embryo(s) during the culture while allowing their retrieval (Fig. 2). The possibility to recover the embryos from microfluidic devices has been previously deemed essential in order to test for full-term development,2 but has not yet been demonstrated. The V-shaped structures implemented at the inlet of the chamber present a 70 μm spacing, which is slightly smaller than the size of mouse embryos (ca. 80 μm) to enable their passage when a mild flow is applied, while preventing them from moving out of the chamber in the absence of flow. All microfluidic structures are fabricated from polydimethylsiloxane (PDMS), which has been validated for its inertness towards embryos (ESI SI-1†), and bonded to a glass substrate. | ||
| Fig. 2 Microfluidic device for embryo culture. Picture of the device fabricated from PDMS prior to bonding on a glass substrate: the device includes a nL volume chamber in which embryos are cultured. Embryos are introduced from the inlet reservoir, guided in the chamber (V-shaped structure; 70 μm spacing; top right) and trapped therein with the help of grids (20 μm spacing; bottom right). All scale bars represent 200 μm. | ||
Flow characterization
Determination of flow settings inside the microfluidic chamber is essential to characterize culture conditions in the proposed device. Flows are created in the device using “passive pumping”,20 which relies on the deposition of round droplets of different sizes on the inlet and outlet reservoirs, without the need for complex fluidic connections. Flows in the chamber are characterized as a function of the chamber dimensions with the help of 3 μm beads added in the liquid. Fig. 3A illustrates the flow profile observed in a 270 nL chamber when a 1.5 μL droplet is applied in the inlet and a 10 μL droplet on the outlet. A flow gradient is found along the outlet grids, where embryos are generally located. The maximal flow value is estimated to be around 64.8 μL h−1 in absence of embryos, decreasing when embryos are present in the chamber (embryos reduce flow as they occupy the chamber close to the outer microchannel; e.g., 50% reduction for 20 embryos in a 270 nL chamber). | ||
| Fig. 3 Fluidic characterization of the microfluidic device. (A) Flow profile in a 270 nL chamber using passive pumping20 after supplementation of the liquid with microbeads (pumping droplet of 1.5 μL). Flow values from right to left: 3.9 μL h−1, 6.5 μL h−1, 16.2 μL h−1, 32.4 μL h−1. (B and C) Development of a medium refreshment protocol. The device is firstly filled with a solution of fluorescein in PBS (green), and a rhodamine B solution (red) in PBS is pumped using the passive pumping technique in the chamber using 1.5 μL droplets (1.5 μL = 1 pumping cycle) until no green fluorescence is detected. (B) Pictures showing the process for a 270 nL chamber after 0, 2, 4, and 7 pumping cycles. All scale bars represent 500 μm. (C) Table showing the number of pumping cycles necessary to fully refresh 30 and 270 nL chambers. | ||
Once the flow settings have been characterized, the number of pumping cycles (using 1.5 μL droplets) required to fully refresh the content of the chamber is determined using fluorescent dyes (Fig. 3B): a rhodamine solution is pumped in a chamber previously filled with a fluorescein solution (Fig. 3B). Typically, 3 or 7 cycles are required to completely replace the culture medium in 30 nL and 270 nL chambers, respectively, as indicated in Fig. 3C. The presence of embryo(s) in the culture chamber is found to have a negligible influence on this medium refreshment protocol (data not shown).
Pre-implantation development characterization in microfluidic environment and conventional droplets
In mouse development, the physiological time window for blastocyst formation (i.e., the embryonic stage in preparation for implantation in the uterus) ranges from 3.5 to 4.5 dpc in vivo, with cavity (blastocoel) formation starting at 72 h post fertilization. Mouse embryos cultured in the microfluidic systems develop significantly faster than in droplets, as measured by the earlier appearance of a cavity and its larger size at 3.5 dpc (Table 1). This result is independent of the chamber volume and applies to embryos cultured as groups of 5, or individually. For pools of 20 embryos, the chamber volume impacts on the development rate: blastocyst formation at 3.5 dpc is delayed for embryos cultured in 30 nL chambers (only 13.3% blastocysts), while in 270 nL chambers the blastocyst rate (54.7%) is only slightly lower than for smaller embryo groups. Interestingly, in droplets, none of the embryos cultured individually reaches the blastocyst stage at 3.5 dpc, while 72.3% and 63.1% of those placed in 30 and 270 nL chambers, respectively, already present a cavity (see Fig. 4).| Culture conditions | 3.5 dpc | 4.5 dpc | ||||||
|---|---|---|---|---|---|---|---|---|
| Vol. | Flow | Embryo nr. | Starting nr.a | Blast. nr. | Blast. rate (%) | Starting nr.a | Blast. nr. | Blast. rate (%) |
| aUnfertilized embryos (i.e., one-cell at 2.5 dpc) have been removed from the calculations, b,c,d,e,fvalues sharing one superscript letter are not significantly different. | ||||||||
| 30 nL | Static | 1 | 119 | 86 | 72.3 ± 4.1b | 36 | 35 | 97.2 ± 2.7f |
| 5 | 132 | 97 | 73.5 ± 3.8b | 37 | 34 | 91.9 ± 4.5f | ||
| 20 | 60 | 8 | 13.3 ± 4.4c | 76 | 69 | 90.8 ± 3.3f | ||
| Dyn. | 5 | 81 | 59 | 72.8 ± 4.9b | 18 | 17 | 94.4 ± 5.4f | |
| 270 nL | Static | 1 | 38 | 24 | 63.1 ± 7.8b | 16 | 15 | 93.7 ± 6.1f |
| 5 | 93 | 71 | 76.3 ± 4.4b | 35 | 32 | 91.4 ± 4.7f | ||
| 20 | 53 | 29 | 54.7 ± 6.8b | 53 | 48 | 90.6 ± 4.0f | ||
| Dyn. | 5 | 78 | 55 | 70.5 ± 5.1b | 20 | 20 | 100e,f | |
| 5 μL | Static | 1 | 28 | 0 | 0c | 47 | 14 | 29.8 ± 6.4b |
| 5 | 27 | 2 | 7.4 ± 5.0c | 27 | 18 | 66.6 ± 9.1c,d | ||
| 20 | 79 | 16 | 20.2 ± 4.5c | 79 | 58 | 73.4 ± 5.0d,e | ||
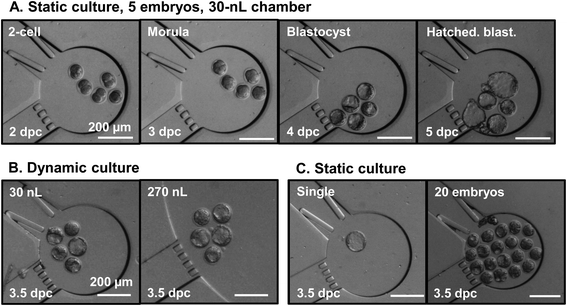 | ||
| Fig. 4 Microscopy pictures of embryos cultured in nL chambers under various conditions. Top row (A): time-lapse imaging of the complete development of a pool of 5 mouse embryos in a 30 nL chamber under static conditions, from the 2-cell stage (2 dpc) until the expanded blastocyst stage (4.5 dpc). Bottom row, from left to right: (B) pools of 5 embryos cultured under dynamic conditions (medium refreshment at 3 dpc) in 30 and 270 nL chambers; (C) single embryo and pools of 20 embryos cultured under static conditions in 30 nL chambers. All pictures in (B) and (C) are taken at 3.5 dpc. All scale bars represent 200 μm. | ||
At 4.5 dpc, the microfluidic environment significantly increases the chance of mouse embryos to become blastocysts compared to droplets (Table 1). All microfluidics conditions yield blastocyst rates above 90%, compared to significantly lower rates in drops (29.8, 66.6 and 73.4% for 1, 5, and 20 embryos, respectively). The results concerning drop culture of mouse embryos are in full agreement with previously published data. For instance, Kato and Tsunoda reported blastocyst rates of 38 and 63% for the culture of single embryos and pools of 10 embryos, respectively, in 10 μL microdrops under similar conditions.26 Furthermore, the development of embryos cultured as pools of 20 in 30 nL chambers is restored at 4.5 dpc (90.8%) with rates comparable to all microfluidic conditions. The widely reported group size effect in pre-implantation development,26–29 which is also found here in the droplet culture, is not observed in the microfluidic environment.
As previously reported for various mammalian species, the total cell number is considered as a good indicator for the quality of pre-implantation embryos, as well as a predictor for good development,30–33 with morphological blastocyst scoring methods correlating with cell numbers in the human.34 In the present study, within the same experiment, single embryo culture produces a significant increase in total blastocyst cell numbers at 4.5 dpc, in both 30 nL (78 ± 23 cells, n = 7; p = 0.0002) and 270 nL (54 ± 10 cells, n = 5; p = 0.009) chambers compared to droplets (34 ± 7 cells, n = 10). Most importantly, total cell counts for single embryo culture in a microfluidic environment are comparable to those of embryos cultured in pools of 20 in droplets (30 nL p = 0.57; 270 nL p = 0.05). Also in droplets, total cell numbers are significantly higher for embryos cultured in pools of 20 (72 ± 13 cells, n = 8) than for single culture (34 ± 7 cells, n = 10; p = 6.4 × 10−6), as already widely reported in the literature.26,35
Full-term development
Mouse embryo culture has already been reported using microdevices.12–15,17,18 Most of these studies concur in indicating that a confined environment as found in microfluidic systems gives developmental advantage during early pre-implantation, compared to the conventional droplet approach.13,14 However, the demonstration of pre-implantation development alone is not sufficient to prove that an in vitro culture system does not harm the embryos.22 Therefore, we examine the full-term development of mouse embryos cultured throughout the pre-implantation period in a confined nL volume chamber.Single embryo culture in droplets supports lower birth rates (20%) compared to group culture (37.5%); these data are in good agreement with previously published reports.26,28,35 On the contrary, full-term development of individually cultured mouse embryos is enhanced in a microfluidic confined environment (30 nL: 33.3%; 270 nL: 28.9%) compared to drops (20%). Furthermore, for culture in microfluidic devices, the chamber capacity (30 and 270 nL) has no influence on the embryo developmental competence, while the embryo number does. Groups of 20 embryos in chambers give the highest birth rates (30 nL: 42.8%; 270 nL: 45.4%) surpassing standard (i.e., droplet) culture conditions (37.5%), while surprisingly for groups of 5 embryos the lowest birth rates are observed (30 nL: 19.1%; 270 nL: 15.2%). These unexpected variations in the developmental rates as a function of the group size raise interesting biological questions for future studies. A possibility is that a fine balance exists between the amount of essential (e.g., growth factors) and deleterious molecules released by the embryos themselves, as well as the availability of the right physical conditions (such as availability of nutrients and oxygen).
Most importantly, our data show that mouse embryos can develop in a microfluidic environment under the prescriptions of human ART, i.e., single embryo culture. So far, various attempts to culture single mouse embryos in droplets have led to impaired development and a decrease in cell numbers,26,35 both being restored upon supplementation of specific growth factors in the medium.28 In the present study, we demonstrate for the first time viable full-term development of embryos cultured individually in a confined microfluidic environment, with growth rates and cell numbers comparable to embryos cultured in groups in either microfluidic chambers or conventional droplets (Table 2). The confinement found in the chambers is likely to play a crucial role for single embryo development, namely through the local accumulation of growth factors secreted by the embryos25 that promote their development28 and the formation of gradients of nutrients in the embryo environment, as found in vivo.1,36 The microfluidic system presented here provides a valuable platform for the future clarification of all the above hypotheses.
| Culture conditions | 3.5 dpc | ||||
|---|---|---|---|---|---|
| Vol. | Flow | Embryo nr. | Starting nr. | Pups nr. | Birth rate (%) |
| a,b,c,dValues sharing one superscript letter are not significantly different. | |||||
| 30 nL | Static | 1 | 36 | 12 | 33.3 ± 7.9c,d |
| 5 | 47 | 9 | 19.1 ± 5.7a,d | ||
| 20 | 28 | 12 | 42.8 ± 9.4c | ||
| Dyn. | 5 | 39 | 11 | 28.2 ± 7.2a,c,d | |
| 270 nL | Static | 1 | 45 | 13 | 28.9 ± 6.8a,c,d |
| 5 | 59 | 9 | 15.2 ± 4.7d | ||
| 20 | 22 | 10 | 45.4 ± 10.6b,c | ||
| Dyn. | 5 | 34 | 10 | 29.4 ± 7.8b,d | |
| 5 μL | Static | 1 | 20 | 4 | 20.0 ± 8.9b,d |
| 5 | 40 | 15 | 37.5 ± 7.7a,b,c | ||
| 20 | 40 | 15 | 37.5 ± 7.7a,b,c | ||
| 30 + 270 nL | Static | 5 | 106 | 18 | 16.9 ± 3.6a |
| Dyn. | 73 | 21 | 28.8 ± 5.3b | ||
| 5 μL | Static | 5 | 40 | 15 | 37.5 ± 7.7b |
Medium perfusion and embryo developmental competence
Medium refreshment during culture is generally considered to promote embryo development.16 Medium substitution is also a common procedure in many ART laboratories, as different media compositions are believed to bring specific beneficial effects at different pre-implantation stages. In other cases, medium is simply refreshed during the culture. In our study, this parameter is tested on groups of 5 embryos in microfluidic chambers. Refreshing the medium at 3 dpc (as for ART sequential media protocols) has no influence on embryo pre-implantation rates (Table 1), most probably as the medium is replenished only shortly before blastocyst rates are assessed (3.5 dpc). Interestingly, medium refreshment promotes full-term development over static conditions (Table 2), as shown by the significant increment in birth rates (from 16.9% to 28.8% for microfluidic systems without and with medium refreshment, respectively).Hickman et al. showed impaired development for embryos exposed to a continuous but low flow-rate (0.1–0.5 μL h−1),12 and a 1.2 dyne cm−2 shear stress has been reported as lethal for mouse embryos.37 On the contrary, pulsatile delivery of medium has recently been demonstrated to promote embryo development.16 Here, embryo development seems to benefit from the transient (1–2 min) but much higher flow (4–50 μL h−1; maximum shear stress ca. 0.17 dyne cm−2) which is created upon punctual medium refreshment using passive pumping.20 We hypothesize this positive impact originates from the mild and punctual mechanical stimulation exerted on the embryos. Interestingly, this emulates the conditions the embryos experience in vivo throughout their first days of development, particularly when they travel in the oviduct from the ovary to the uterus.16
Conclusion
This study is the first comprehensive account that a nanoliter volume not only can support pre-implantation development in vitro, but it can also preserve the post-implantation ability, resulting in full-term development after blastocyst transfer to the uterus. The system offers the possibility to non-invasively assess the impact of culture conditions on embryo growth. In particular, this set of experiments has revealed three notable features: first, microfluidic chambers support full-term development of mouse embryos with birth rates comparable to droplet culture; second, medium refreshment in microfluidic systems (tested here on groups of 5 embryos) has a significant impact on embryo full-term development; third, and most importantly, single embryo culture in a microfluidic environment gives birth rates and cell numbers comparable to group culture in droplets. This last key result – that mouse embryos develop further to term when cultured in a confined environment under the prescription of human ART, i.e., single embryo culture – is a major step for the field of ART. The reason why post-implantation rates are here – in the mouse – varying across group sizes while being independent of the microfluidic volume remains to be investigated. However, it is known that mouse blastocyst rates are not predictive of foetal rates, whereas blastocyst cell numbers are.32 The most parsimonious explanation for the lack of difference in full development is that the minimum viable number of cells was secured in all of our experiments owing to the fact that mouse oocytes were fertilized in vivo, i.e., under best conditions. It will be interesting also to see the effect of microfluidic embryo culture on embryos fertilized by other ART methods such as intracytoplasmic sperm injection (ICSI), which is an invasive technique predicted to affect blastocyst cell numbers. Considering the historical records in which single embryo culture – which is praxis in human ART – has always proven to be problematic for the mouse,26–28 our present achievements narrow the gap between the mouse as a model for human embryo development and human ART, by establishing that mouse embryos can be cultured in a way that matches human embryos (i.e., at the single embryo level). In parallel, the milestone achieved in this study also enables re-thinking embryo culture in microfluidic platforms comprising individual chambers for parallel single embryo culture. This single embryo approach suppresses risks of low quality embryos negatively influencing nearby fellow embryos,38 and facilitates embryo tracking during the culture while enabling to tailor culture conditions for each embryo. Importantly, physiological data can be collected at the single embryo level using dedicated sensors and analytical tools implemented in nL chambers or microdevices.39–43 Such data will undoubtedly not only help identifying the physiological requirements of pre-implantation embryos, but also provide new means to monitor embryo growth and assess their quality using more rational criteria than morphology alone,44 as currently done in the clinics. Furthermore, embryo development can simultaneously be followed using time-lapse imaging45 in the microfluidic chambers, e.g., to correlate physiological data and developmental stage. Finally, this combination of single embryo culture in microfluidic systems with in situ characterization scheme is highly valuable to screen physical and chemical culture parameters, as undertaken in the present study, towards the determination of an optimal in vitro culture protocol.Acknowledgements
The authors would like to thank the technicians of the BIOS group for their precious help, as well as the animal caretakers at the MPI Münster, for animal work. This research is funded by the bilateral project NWO-DFG DN 63-258 and the DFG BO 2540/4-1 (MB).References
- G. Vajta, L. Rienzi, A. Cobo and J. Yovich, Reprod. BioMed. Online, 2010, 20, 453–469 CrossRef PubMed.
- J. E. Swain and G. D. Smith, Hum. Reprod. Update, 2011, 17, 541–557 CrossRef CAS PubMed.
- G. A. Thouas, G. M. Jones and A. O. Trounson, Reproduction, 2003, 126, 161–169 CrossRef CAS PubMed.
- G. Vajta, T. Korosi, Y. Du, K. Nakata, S. Ieda, M. Kuwayama and Z. P. Nagy, Reprod. BioMed. Online, 2008, 17, 73–81 CrossRef.
- G. Vajta, T. T. Peura, P. Holm, K. Paldi, T. Greve, A. O. Trounson and H. Callesen, Mol. Reprod. Dev., 2000, 55, 256–264 CrossRef CAS.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- T. M. Keenan and A. Folch, Lab Chip, 2008, 8, 34–57 RSC.
- S. Le Gac and A. van den Berg, Trends Biotechnol., 2010, 28, 55–62 CrossRef CAS PubMed.
- E. M. Lucchetta, J. H. Lee, L. A. Fu, N. H. Patel and R. F. Ismagilov, Nature, 2005, 434, 1134–1138 CrossRef CAS PubMed.
- H. Hwang and H. Lu, Biotechnol. J., 2013, 8, 192–205 CrossRef CAS PubMed.
- T. J. Levario, M. Zhan, B. Lim, S. Y. Shvartsman and H. Lu, Nat. Protoc., 2013, 8, 721–736 CrossRef PubMed.
- D. L. Hickman, D. J. Beebe, S. L. Rodriguez-Zas and M. B. Wheeler, Comp. Med., 2002, 52, 122–126 CAS.
- S. Raty, E. M. Walters, J. Davis, H. Zeringue, D. J. Beebe, S. L. Rodriguez-Zas and M. B. Wheeler, Lab Chip, 2004, 4, 186–190 RSC.
- J. Melin, A. Lee, K. Foygel, D. E. Leong, S. R. Quake and M. W. M. Yao, Dev. Dyn., 2009, 238, 950–955 CrossRef PubMed.
- R. Ma, L. Xie, C. Han, K. Su, T. Qiu, L. Wang, G. Huang, W. Xing, J. Qao, J. Wang and J. Cheng, Anal. Chem., 2011, 83, 2964–2970 CrossRef CAS PubMed.
- Y. S. Heo, L. M. Cabrera, C. L. Bormann, C. T. Shah, S. Takayama and G. D. Smith, Hum. Reprod., 2010, 25, 613–622 CrossRef CAS PubMed.
- C. Han, Q. Zhang, R. Ma, L. Xie, T. Qiu, L. Wang, K. Mitchelson, J. Wang, G. Huang, J. Qiao and J. Cheng, Lab Chip, 2010, 10, 2848–2854 RSC.
- M. S. Kim, C. Y. Bae, G. Wee, Y.-M. Han and J.-K. Park, Electrophoresis, 2009, 30, 3276–3282 CrossRef CAS PubMed.
- X. M. Zhao, Y. N. Xia and G. M. Whitesides, J. Mater. Chem., 1997, 7, 1069–1074 RSC.
- G. M. Walker and D. J. Beebe, Lab Chip, 2002, 2, 131–134 RSC.
- F. M. Cavaleri, S. T. Balbach, L. Gentile, A. Jauch, B. Bbhm-Steuer, Y.-M. Han, H. R. Schbler and M. Boiani, Mech. Dev., 2008, 125, 153–166 CrossRef CAS PubMed.
- C. Schwarzer, T. C. Esteves, M. J. Arauzo-Bravo, S. Le Gac, V. Nordhoff, S. Schlatt and M. Boiani, Hum. Reprod., 2012, 27, 2627–2640 CrossRef CAS PubMed.
- N. Desai, D. Kinzer, A. Loeb and J. Goldfarb, Hum. Reprod., 1997, 12, 328–335 CrossRef CAS PubMed.
- W. S. A. Wun, C. C. C. Wun and G. M. Grunert, J. Assisted Reprod. Genet., 1994, 11, 303–307 CrossRef CAS.
- H. M. Yu, C. M. Alexander and D. J. Beebe, Lab Chip, 2007, 7, 726–730 RSC.
- Y. Kato and Y. Tsunoda, Theriogenology, 1994, 41, 1315–1322 CrossRef CAS.
- R. S. Canseco, A. E. T. Sparks, R. E. Pearson and F. C. Gwazdauskas, J. Assisted Reprod. Genet., 1992, 9, 454–457 CrossRef CAS.
- B. C. Paria and S. K. Dey, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 4756–4760 CrossRef CAS.
- M. Lane and D. K. Gardner, Hum. Reprod., 1992, 7, 558–562 CAS.
- T. P. Fleming, Dev. Biol., 1987, 119, 520–531 CrossRef CAS.
- V. E. Papaioannou and K. M. Ebert, Development, 1988, 102, 793–803 CAS.
- M. Lane and D. K. Gardner, J. Reprod. Fertil., 1997, 109, 153–164 CrossRef CAS PubMed.
- S. Iwasaki, N. Yoshiba, H. Ushijima, S. Watanabe and T. Nakahara, J. Reprod. Fertil., 1990, 90, 279–284 CrossRef CAS PubMed.
- K. Matsuura, N. Hayashi, C. Takiue, R. Hirata, T. Habara and K. Naruse, Fertil. Steril., 2010, 94, 1135–1137 CrossRef PubMed.
- B. D. Bavister, Theriogenology, 1988, 29, 143–154 CrossRef.
- B. D. Bavister, Hum. Reprod. Update, 1995, 1, 91–148 CrossRef CAS PubMed.
- Y. F. Xie, F. F. Wang, W. J. Zhong, E. Puscheck, H. L. Shen and D. A. Rappolee, Biol. Reprod., 2006, 75, 45–55 CrossRef CAS PubMed.
- R. E. Spindler and D. E. Wildt, Biol. Reprod., 2002, 66, 167–173 CrossRef CAS.
- Y. S. Heo, L. M. Cabrera, C. L. Bormann, G. D. Smith and S. Takayama, Lab Chip, 2012, 12, 2240–2246 RSC.
- J. P. Urbanski, M. T. Johnson, D. D. Craig, D. L. Potter, D. K. Gardner and T. Thorsen, Anal. Chem., 2008, 80, 6500–6507 CrossRef CAS PubMed.
- K. Komori, S. Fujii, K. Montagne, H. Nakamura, H. Kimura, K. Otake, T. Fujii and Y. Sakai, Sens. Actuators, B, 2012, 162, 278–283 CrossRef CAS PubMed.
- C. C. Wu, T. Saito, T. Yasukawa, H. Shiku, H. Abe, H. Hoshi and T. Matsue, Sens. Actuators, B, 2007, 125, 680–687 CrossRef CAS PubMed.
- C. O'Donovan, E. Twomey, J. Alderman, T. Moore and D. Papkovsky, Lab Chip, 2006, 6, 1438–1444 RSC.
- A. T. Kjellberg, P. Carlsson and C. Bergh, Hum. Reprod., 2006, 21, 210–216 CrossRef PubMed.
- C. C. Wong, K. E. Loewke, N. L. Bossert, B. Behr, C. J. De Jonge, T. M. Baer and R. A. R. Pera, Nat. Biotechnol., 2010, 28, 1115–U1199 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra44453h |
| This journal is © The Royal Society of Chemistry 2013 |