 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
B-TUD-1: a versatile mesoporous catalyst†
Adeline Ranoux, Kristina Djanashvili, Isabel W. C. E. Arends and Ulf Hanefeld*
Technische Universiteit Delft, Department of Biotechnology, Gebouw voor Scheikunde, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: u.hanefeld@tudelft.nl; Fax: +31 (0) 15 2781415
First published on 5th September 2013
Abstract
Novel amorphous mesoporous borosilicate, B-TUD-1, was prepared to test its performance for different sustainable reactions. The structure of the material, the effective incorporation of boron into the framework as well as the nature of incorporated boron were verified by N2-sorption, XRD, ICP-OES, TEM, NH3-desorption, MAS NMR and FTIR. The potential of these materials as catalysts was tested in two reactions under different conditions. They showed an apparently good activity and recycling potential for HMF (5-hydroxymethyl-furfural) synthesis. However leaching of boron occurred during this aqueous reaction. We could demonstrate that the leached boron was actually the catalyst of the reaction. In a second reaction, this time in organic solvent, the B-TUD-1 materials were tested for the Prins cyclisation of citronellal. A very promising activity was obtained. The catalyst could be recycled and no boron leached, as demonstrated by hot filtration experiments.
Introduction
Heterogeneous catalysts were widely studied during the last few years, in particular for the development of sustainable processes. Indeed, while representing powerful catalysts, they have the additional advantage that they are easily recyclable and thus lead to significant reductions of waste. In this context, TUD-1 materials were developed.The special properties of mesoporous TUD-1, such as its three dimensional pore structure with a good accessibility for substrates and products, tuneable pore properties,1,2 and a cost-effective one-pot, surfactant-free synthesis by the sol–gel method, have made it a versatile heterogeneous catalyst for many interesting synthesis applications.3 TUD-1 has been successfully functionalized with numerous incorporated metals (Al, Ti, Co, Fe, V, Cr, Mo, Hf, Ga, Ni, Mn, Zr and Cu) aiming at a variety of catalytic properties.4–29 Surprisingly, the incorporation of B, in the same group as Al, was never investigated.
Boron derivatives have proven to be efficient catalysts for a large range of reactions (reduction, Diels–Alder, aldol reactions).30,31 Boric acid itself is a good Lewis acid for esterifications,32 amidifications33 and dehydrations of polyols.34–36
Both TUD-1 materials and boric acid have been demonstrated as excellent catalysts for sustainable reactions, conversion of renewable materials and synthesis of platform chemicals (for example, boric acid catalysed synthesis of HMF37 or Hf-TUD-1 catalysed conversion of glycerol to solketal).14
Previously other boron-incorporated zeolites and mesoporous materials (Beta- and ZSM-5 zeolites, SBA-15, MCM-41)38–49 have been reported. Key examples are the B-MCM-41-catalysed Beckmann rearrangement of cyclohexanone oxime44 permitting the efficient synthesis of caprolactam, and, the B-MCM-41-catalysed three component Strecker reaction also leading to the formation of α-aminonitriles,50 intermediates for the synthesis of α-aminoacids or N-containing heterocycles. Boron is herewith a promising, cheap and environmentally benign element that imparts activity to mesoporous materials. In this paper a novel approach towards the synthesis of heterogeneous mesoporous B-catalyst is presented, B-TUD-1. Combining the advantageous properties of the TUD-1 materials (notably their 3D structure suppressing diffusion limitation of the substrates) and excellent catalysis efficiency of the boron derivatives, the obtained B-TUD-1 catalysts should present new versatile catalytic properties. We present here not only the preparation of the catalyst and its full characterization, and additionally performance tests for two important, sustainable reactions, the HMF synthesis, and, the Prins cyclisation of citronellal.
Results and discussion
Preparation of the material
The M-TUD-1 materials (Al and B) were prepared by the sol–gel method using triethanolamine as a complexing agent following the protocols described earlier.9 The M was included in the form of the isopropoxide M(iPrO)3.The use of triethanolamine permits to avoid the use of any surfactants or liquid crystal templates. Complexing with the metal triethanolamine leads to the formation of the mesopores (Fig. 1). Initially the active metal is complexed to form an atrane.51,52 Then these atranes complex together with free triethanolamine creating the framework of the material. During gelation and also after calcination and consequent removal of organic compounds, the metal is incorporated into the framework of the material as oxide species.3,4 This is also known as the atrane route.53 The formation of the boratranes has been demonstrated as to be a set of complex equilibria between the different coordination forms. Several stable boratranes with three six-membered rings where the boron has a tetrahedral structure could be characterised.54–56 Another interesting point is that by the use of a similar method introducing surfactants, ordered mesoporous materials could be obtained (for example SBA-15 57) in contrast to the amorphous, mesoporous TUD-1 materials.
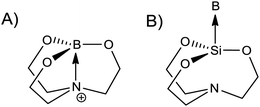 | ||
| Fig. 1 Atranes complexes, triethanolamine as (A) metal complexing agent and (B) templating agent. | ||
Different ratios of M were introduced, Si/Al 25 (Al-TUD-1 25), and Si/B 50, 25 and 10 (B-TUD-1 50, 25 and 10 resp.). A hydrothermal treatment of 6 h at 180 °C was applied that has defined the size of the pores and the mesoporous structure.1 Subsequent calcination at 600 °C for 10 hours led to the final material.
Characterization: a classical mesoporous catalyst
The ratio of incorporated boron was verified by elemental analysis by inductively coupled plasma atomic emission spectroscopy (ICP-OES). The mesoporous character of the materials was confirmed by X-ray diffraction (XRD), N2 sorption and high-resolution transmission electron micrographs (TEM). The acid sites were characterised by temperature-programmed desorption of NH3. Finally, the incorporation of boron was verified by XRD at small angles and by MAS NMR. The latter permitted also to characterise the nature of the boron species incorporated into the mesoporous framework.Results of the elemental analysis (ICP-OES) and the porosity measurements are given in Table 1. The elemental analysis reveals Si/B ratios of the calcined catalysts very close to the actual added amount. ≥95% of the added boron compounds during the preparation of the gel was incorporated. This demonstrates the good predictability of the preparation method, already observed with other M-TUD-1 materials.8,58
According to Jansen et al.,1,2 the pore size of TUD-1 material is tuneable and can be controlled by the time of the hydrothermal treatment. After a treatment of 6 h, the expected average pore size was between 6 and 8 nm. The measured pore diameters correspond to the theoretical values and are slightly larger than those for classical M-TUD-1 (average around 4.0 nm)3 and close to other boron containing mesoporous silicates (for example, B-MCM-41, average around 4.5 nm, B-SBA-15, average around 6.5 nm).43,46,47 The surface area of B-TUD-1 increases upon increasing of the metal loading. At the same time, the average pore diameter decreases in line with the results for other M-TUD-1 materials3 and the boron substituted mesoporous SBA-15.43
The N2 adsorption and desorption isotherms of the B-TUD-1 50 and 25 (Fig. 2) reveal a typical “type IV” hysteresis loop, with a large uptake of nitrogen at relative pressures between 0.5 and 0.9 due to capillary condensation in the mesopores. A plateau at relative pressures of above 0.9 p/p0 indicates the absence of large mesopores or macropores. The B-TUD-1 10 isotherm (Fig. 2) differs slightly by the fact that it shows a lower uptake in the first phase, possibly related to a lower mesoporosity. The material has the lowest pore diameter and micropore volume (Table 1). This might result from the intensified interaction of the B atoms with the silica structure as it has been observed for the Zr-TUD-1 material.58
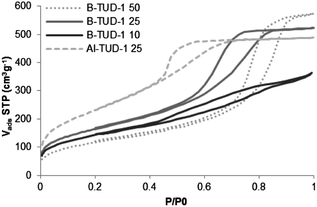 | ||
| Fig. 2 N2 adsorption and desorption isotherms at 77 K. | ||
The X-ray diffractograms of the materials (Fig. 3) show a broad intense peak at low angles around 0.5–1 2θ° in line with the mesostructured character of these materials. No correlation could be established between the peak intensity and the Si/B ratio.
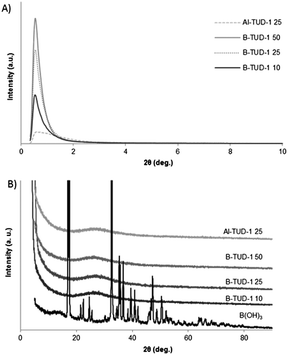 | ||
| Fig. 3 Powder XRD diffraction patterns of calcined B-TUD-1 (A) at small angles (0.5 to 10 deg.); (B) at higher angles (4 to 90 deg.). | ||
The complete XRD spectra of B-TUD-1 10, 25 and 50, as well as that of Al-TUD-1 25 for comparison, were recorded. No crystalline structure was observed in any case. A spectrum of boric acid was measured as a reference (multiple peaks, characteristic of this crystalline product). None of the characteristic signals of boric acid could be detected in the B-TUD-1 spectra, not even at a Si/B ratio of 10. This is quite remarkable compared to other M-TUD-1 materials. It indicates a complete framework incorporation even at the highest metal loading, where most of the M-TUD-1's contain a certain percentage of metal oxides visible on TEM pictures.3,8 Complete boron incorporation at high loading has also been reported for other boron-containing zeolites38,45 or mesoporous silicates.43,44
The TEM pictures (Fig. 4) show “worm/sponge-like” structures typical for amorphous material and TUD-1 samples.1,3 This confirms the mesoporous structure of the material. Furthermore, no crystalline boron particles could be observed even at higher loading confirming the incorporation of the boron species. Therefore, the material appeared completely amorphous, as no ordered pores or channels were visible, and as no boron (or aluminium) oxide could be detected (Fig. 3 and 4).
 | ||
| Fig. 4 TEM pictures of B-TUD-1 10 revealing the mesostructure of the catalysts (scale in nm) (A) 10 nm, (B) 20 nm, (C) 50 nm. | ||
A temperature-programmed desorption (TPD) profile (from 100 to 500 °C) of ammonia for different B- and Al-TUD-1 samples (Fig. 5) was measured. For all the boron-incorporated materials two peaks were observed, an intense peak at 150 °C corresponding to weak acids, and a broad peak with low intensity at around 400–500 °C, corresponding to strong acid sites (Table 2). The medium acid sites (temperature around 250–300 °C) are hardly present. The intensity of the peaks (especially for the weak acid sites) increases with the B loading (i.e. decreasing Si/B ratio) as well as the total acidity. In the case of Al-TUD-1 25, we could observe one broad band from 100 to 350 °C corresponding to weak and medium acids, more classically observed with M-TUD-1 catalysts.11
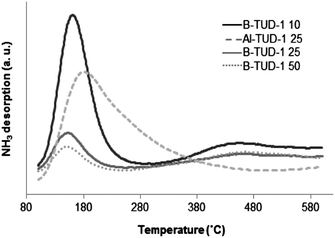 | ||
| Fig. 5 Temperature-programmed desorption (TPD) profile of ammonia for various Al- and B-TUD-1 samples. | ||
| Catalyst | Total acidity (mmol NH3 g−1) | Temp (°C) | Quantity (μmol NH3 g−1) |
|---|---|---|---|
| B-TUD-1 Si/B 10/1 | 0.428 | 161 | 273.3 |
| 295 | 0.9 | ||
| 459 | 152.4 | ||
| B-TUD-1 Si/B 25/1 | 0.195 | 152 | 71.7 |
| 473 | 115.8 | ||
| B-TUD-1 Si/B 50/1 | 0.147 | 148 | 35.9 |
| 107.9 | |||
| Al-TUD-1 Si/Al 25/1 | 0.372 | 179 (max) | 372 |
The solid-state 11B MAS NMR (Fig. 6) showed three main peaks for the B-TUD-1 samples, corresponding to the earlier results for the boron containing mesoporous silicates as MCM-41 (ref. 42) or SBA-15 (ref. 43) (and to the results obtained for many zeolites39,40,49). The first peak at around −22.9 ppm, sharp and narrow, corresponds to the 4-coordinated tetrahedral boron (mainly, 2-coordinated also possible,43 symmetric form). The two other broader bands from −11.5 ppm to −2.5 ppm correspond to different trigonal forms of boron species with different degrees of coordination to silica (in line with literature39,40,42,49), namely 3-coordinated (11.5 ppm), and trigonal B with a lower coordination number (structures in Fig. 7). With a decrease in boron loading, i.e. higher Si/B ratio, relatively more tetrahedral boron is observed. It is however important to notice the dynamic nature of these incorporated boron species. Diverse equilibria are established between the different species, especially between tetrahedral and trigonal forms, as function of the temperature and media (hydration, presence of cations),45,49 explaining the indistinguishable broad peaks observed in the NMR spectra. For this reason, the nature of the acid sites is also dynamic. Due to this dynamic equilibrium that is also influenced by water, it is not meaningful to determine the ratio of Lewis to Brønsted acid sites as this is dynamic too. Furthermore, the 27Al MAS NMR spectrum of the Al-TUD-1 material was also measured (cf. ESI; Fig. S1†). Two main peaks were observed at 0 and 55 ppm, corresponding to the octa- and tetrahedral aluminium resp., as described in the literature.59–61
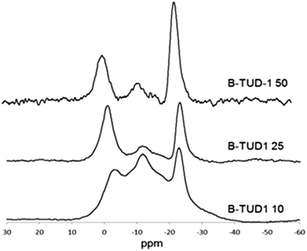 | ||
| Fig. 6 11B MAS NMR of B-TUD-1 catalysts (25, 10) referenced to 0.1 M aq. B(OH)3 (most of the cited articles use BF3(OEt)2 as reference leading to a shift of the signals of 19.8 ppm). | ||
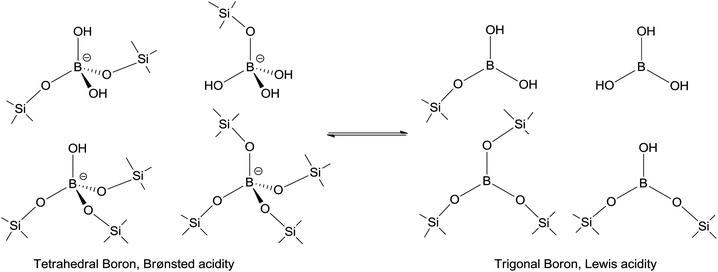 | ||
| Fig. 7 Possible tetrahedral and trigonal structures of boron incorporated in TUD-1 framework. | ||
The presence of tetrahedral boron species in B-TUD-1 materials was confirmed by FTIR as a shoulder at 920–930 cm−1 (mainly visible for B-TUD-1 10, Fig. 8). Another peak characteristic for the presence of trigonal boron can be identified at 1395 cm−1.43,47
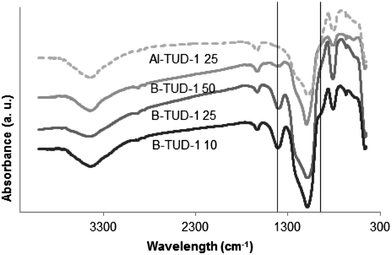 | ||
| Fig. 8 FTIR spectra of B-TUD-1 50, 25, 10 and Al-TUD-1 25. | ||
The intensity of these two peaks, related to the presence of B, decreases in line with the B loading. For comparison, these peaks are absent in the spectra of Al-TUD-1. The absorption bands at 460, 810 and 1070 with a shoulder at 1200 cm−1, present in all the B- and Al-TUD-1, can be assigned to the symmetric and asymmetric stretching of Si–O–Si vibrations of tetrahedral SiO2 units (Fig. 8). Based on these results, we can postulate that B-TUD-1 materials with fully framework incorporated boron were prepared. They contain trigonal boron representing Lewis acidity as well as tetrahedral boron representing Brønsted acidity (Fig. 6 and 7) in a dynamic equilibrium.
Performance test in aqueous or biphasic media: HMF synthesis
HMF, 5-hydroxymethylfurfural, is a biobased platform chemical, obtained from sugars, especially fructose, by acid-catalysed dehydration (Scheme 1).62 However, despite the many studies that have been reported, no real efficient, sustainable and scalable route has been proposed yet.62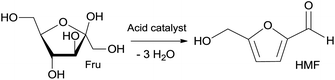 | ||
| Scheme 1 Acid-catalysed dehydration of fructose (Fru) leading to the formation of HMF. | ||
Many Lewis acids,63,64 zeolites and mesoporous materials15,22,65–70 were used as catalysts for this reaction. Because of the numerous advantages (recyclability, easy separation of the product, no corrosion of equipment), heterogeneous catalysts in general have been the main focus.
As Al-TUD-1 60 and boric acid37,71 were already successfully tested for HMF synthesis in biphasic systems or ionic liquids, B-TUD-1 materials appear to be promising heterogeneous, thus potentially recyclable, catalysts for HMF production. Indeed, highly stable, TUD-1 materials have been shown to be easily recovered and recycled after reaction by a calcination similar to the one used during the preparation.3
The synthesis of HMF in aqueous media is a challenging reaction. The B-TUD-1 catalysts showed very good activity for this reaction, especially under biphasic conditions with toluene.
In Fig. 9, experimental results are presented. With the B-TUD-1 catalysts under purely aqueous conditions, the conversion and yield appeared limited (same results at 150 °C for 90 min or 4 h, ≤ 20% yield); whereas in biphasic media, at 150 °C for 4 h, 65 to 70% conversion of fructose was observed with a yield between 40 and 45%. Interestingly, these results were similar for all Si/B ratios and very close to what can be obtained under similar conditions with Al-TUD-1.60
Recycling experiments were successfully carried out in biphasic media (Fig. 10). At 190 °C up to 90% conversion, a yield of 60% can be reached after 40 minutes. This can be repeated in three consecutive cycles.
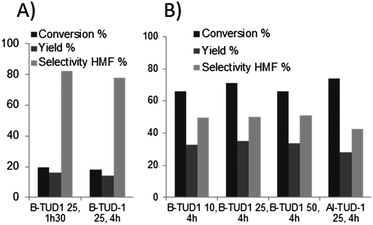 | ||
| Fig. 9 B or Al-TUD-1 catalyzed HMF synthesis in (A) aqueous or (B) biphasic conditions (at 150 °C, 30 wt% fructose, 10 wt% M-TUD-1, toluene/water 3/1). | ||
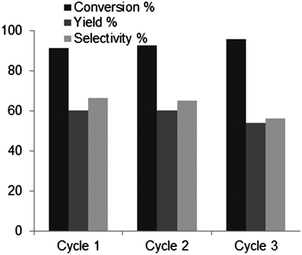 | ||
| Fig. 10 Results of the recycling experiments of B-TUD-1 25 in biphasic conditions (190 °C, 40 min, 30 wt% fructose, 10 wt% M-TUD-1, toluene/water 3/1), conversion of fructose (%), yield of HMF, selectivity to HMF (%). | ||
In the same way, consecutive recycling experiments were carried out in a purely aqueous phase (Fig. 11A). However, the liquid 11B NMR analysis of the reaction media revealed the presence of a broad peak at 0 ppm, corresponding to the presence of boric acid. Furthermore, the elemental analysis of the recycled catalysts after one, two or three reactions revealed an increasing Si/B ratio (from 25 to 85 after 3 reactions, Fig. 11B), thus a decreasing B content on the mesoporous material. The B is probably slowly released during the reaction. Furthermore, a porosity analysis of the used catalysts after calcination showed a clear change in their mesoporous structure (Table 3). Notably a net decrease of the mesoporous surface and pore volume could be observed. The structure is probably affected by the release of B and the hydrolysis of the Si–O–B bonds. Finally, a 11B MAS NMR of the used catalysts (cf. ESI; Fig. S2†) showed a net decrease of the peaks corresponding to tetrahedral 4-coordinated B (sharp peak at −22.9 ppm), but also to the trigonal 3-coordinated B (broad peak at −11.5 ppm). We observed thus under acidic conditions, in line with literature descriptions in the case of different boron containing zeolites,45,49 both the conversion of tetrahedral to trigonal boron, as well as the progressive cleavage of the Si–O–B bridges, leading to a partial deboronation.
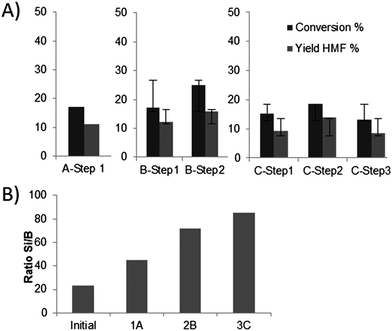 | ||
| Fig. 11 (A) Results of the recycling experiments in aqueous phase (A, 1 reaction cycle; B, 2 reaction cycles/1 recycling step; C, 3 reaction cycles/2 recycling steps; 160 °C; 1 h, 30 wt% fructose, 10 wt% B-TUD-1 25), conversion of fructose (%), yield of HMF; (B) ratio Si/B measured by elemental analysis after one (1A), two (2B), or three (3C) reactions. | ||
This observed leaching, questions the character of the catalysis, heterogeneous ensured by the solid catalyst, or homogeneous ensured by the released boric acid. To clarify this point, a recycling experiment of the aqueous phase of the reaction was carried out (Fig. 12). The catalyst was filtered off after one hour and the reaction was continued with the remaining aqueous phase for an extra hour. In parallel, a reference sample was running, where the catalyst was not removed.
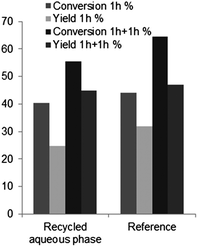 | ||
| Fig. 12 Results of the recycling experiment of the aqueous phase of the reaction medium (175 °C, 1 h, +1 h after filtration or +1 h without filtration for the reference, 30 wt% fructose, 10 wt% B-TUD-1 25), conversion of fructose (%), yield of HMF (%). | ||
The reaction was shown to continue despite the removal of B-TUD-1. Both conversion and yield continued to increase. Moreover, very similar results were obtained with the solid catalyst or after removal of the catalyst (Fig. 12). Our conclusion is that the released boric acid catalyses the HMF formation. Finally, a last experiment (conditions biphasic, 190 °C, 40 min), conducted with a small concentration of boric acid (4 wt%) led to similar results (89% conversion, 59% HMF yield) as the ones observed with the solid catalysts (91% conversion, 60% HMF yield), thus confirming this assertion.
This result with B-TUD-1 can be compared to the stability of Al-TUD-1 under similar conditions. Lima et al.60 showed a good recyclability of Al-TUD-1 under similar reaction conditions. Moreover, our measurement of the 27Al MAS NMR after reaction showed identical spectra to the original calcined sample. This difference in stability can be explained by a smaller size of the boron and its greater dynamic reactivity in the silica framework.
Performance test in organic media: Prins cyclisation
The catalyst performance was also tested in the Prins reaction72–74 (Scheme 2), an important C–C bond forming reaction catalysed by acids.75,76 | ||
| Scheme 2 Intramolecular Prins cyclisation of citronellal. | ||
This intramolecular cyclisation of citronellal, a renewable product, is an essential step in the Takasago synthesis of menthol.77 Industrially, the reaction is catalysed by stoichiometric amounts of solid ZnBr2 in benzene.78 Recently, the implementation of alternative processes using heterogeneous catalysts has attracted considerable attention.11,58,79–89 The Prins reaction was carried out with the different B-TUD-1 (50, 25 and 10) catalysts as well as with Al-TUD-1 25 as a reference (Scheme 3).
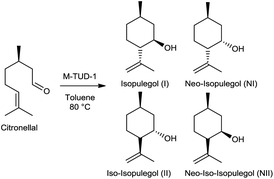 | ||
| Scheme 3 Cyclisation of citronellal to isopulegol and its isomers catalysed by M-TUD-1 material. | ||
The obtained activity of the B-TUD-1 catalysts was quite high (Fig. 13) and was increasing with the amount of incorporated boron (thus with a decreasing Si/B ratio). It should be noticed that the experiments were carried out with industrial grade citronellal containing around 5% isopulegol. As it has previously been shown, the presence of isopulegol partially inhibits the conversion of citronellal. Higher activities can be expected starting from optically pure citronellal.58
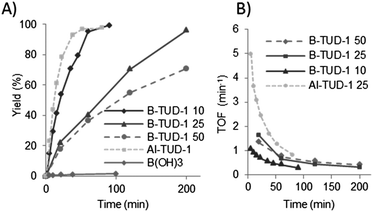 | ||
| Fig. 13 (A) Evolution of the yield of isopulegol formed during the Prins cyclisation catalysed by various B- and Al-TUD-1 catalyst; (B) evolution of the TOF of the catalysts during the reaction (in mol converted product per mol B or Al per min). | ||
We could observe full conversion after one hour for the B-TUD-1 10. This result is very close to the conversion that we obtained via Al-TUD-1 (full conversion after approximately 50 minutes). Full conversion was reached after four hours for B-TUD-1 25, 80% conversion was obtained after 6 h with B-TUD-1 50. Despite an apparent lower activity of the latter two catalysts, the comparison of their turn over frequencies (TOF, Fig. 13B) showed their activity per active site to be actually higher than for B-TUD-1 10.
These results are similar to the results with Zr-TUD-1 100. We can also compare this performance to other examples of zeolite catalysts for this reaction. Similar performance was observed under the exact same conditions with Al-MSU-50 (ref. 83) (88.6% conversion after 30 minutes, and 96.7% after 60 minutes), whereas Zr-incorporated MOF89 only presented a full conversion after a minimum of 4 h at 100 °C for a citronellal/Zr ratio of 10 (in our conditions, with B-TUD-1 10, citronellal/B ratio of 9, with B-TUD-1 25, citronellal/B ratio of 21). Our catalyst thus presents an interesting activity for the Prins cyclisation of citronellal. The product selectivity to isopulegol (all diastereoisomers) for all the catalysts appeared excellent (≥95%, Table 4). The observed diastereoselectivity to isopulegol (vs. its other diastereoisomers) is around 63/37 for all B-containing TUD-1 catalysts (Table 4). This result is close to the measured diastereoselectivity of Al-TUD-1 25 (66/34) and to the values described in literature for Zr-TUD-1 (average 65/35).58 The reduced pore diameter of B-TUD-1 10 does not influence this ratio.58
| Catalyst | Time | Conversion (%) | Selectivity (%) | Diastereoselectivity I/MI/II/NII (%) |
|---|---|---|---|---|
| Al-TUD-1 25 | 60 | 98 | 98 | 66.3/22.6/7.05/0.05 |
| B-TUD-1 10 | 60 | 97 | 98 | 63.2/30.1/5.1/1.6 |
| B-TUD-1 25 | 240 | 96 | 95 | 63.2/30.1/4.8/1.8 |
| B-TUD-1 50 | 360 | 81 | 98 | 62.7/30.2/5.1/2.0 |
A liquid 11B NMR of the reaction medium after filtration of the catalyst did not show the presence of boron compounds, indicating the absence of leaching. Furthermore a porosity measurement of the catalysts after reaction and calcination showed very similar results (Table 3) to the initials catalysts (Table 1). Finally, a hot filtration test was carried out. After 20 min, the reaction medium is sampled, immediately filtered, and left again at 80 °C. This operation is performed on a hot sample to prevent any possible precipitation of diluted compounds.90 After the removal of the solid catalyst, the reaction clearly stopped (Fig. 14), proving the heterogeneous nature of the catalytic activity.
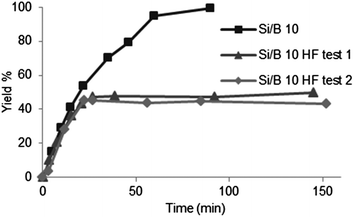 | ||
| Fig. 14 Hot filtration test on B-TUD-1 10 during the Prins cyclisation of citronellal; catalyst filtered after 20 minutes; 80 °C. | ||
Recycling steps were realised with the catalysts B-TUD-1 10 and 25, with or without calcination before the recycling reaction (Fig. 15). Very similar results were obtained with the calcined recycled catalysts, whereas we could observe a net decrease of the activity of the non-calcined samples, probably partially inhibited by the product remaining on the catalyst as described earlier.58
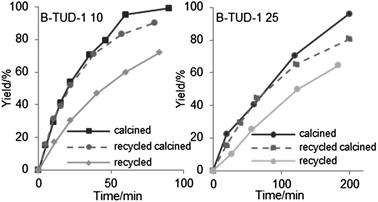 | ||
| Fig. 15 Results of the recycling experiments with or without intermediate calcination of the B-TUD-1 25 and 10 catalysts, yield of isopulegol (%). | ||
Conclusions
We successfully obtained and characterized a new, easy to prepare, three-dimensional and mesoporous acidic B-TUD-1 catalyst. We verified the mesoporous and amorphous character of the material as well as the incorporation of the boron into the framework even at low Si/B ratio. We also characterized the mixed acidity of the material (Brønsted and Lewis acid sites) as well as the different possible structures of the incorporated boron (trigonal and tetrahedral) and their dynamic equilibrium. Catalytic tests showed the efficiency of this material for the catalysis of different sustainable reactions. However, in the case of HMF synthesis, the acidic aqueous reaction conditions caused boron leakage and a change of the structure of the material was observed. On the contrary, B-TUD-1 demonstrated high reactivity as well as reasonable recyclability for the Prins cyclisation of citronellal in toluene. B-TUD-1 is there with a promising heterogeneous acid that catalyses reactions under non-aqueous conditions.Experimental section
Materials and methods
All chemicals were purchased from Aldrich, Janssen or Acros. For the catalysis experiments, the anhydrous solvents and solids were used as received, all other liquids were dried and distilled prior use. M-TUD-1 catalysts were activated in the presence of air at up to 600 °C at a ramp of 1 °C min−1 and heated at 600 °C for 10 h. The experiments were performed in dried glassware and under N2 atmosphere.Catalyst preparation
Catalyst characterization
The volumetric nitrogen adsorption was realised on a Quantachrome Autosorb-6B at 77 K. Prior to the physisorption experiments, samples were degassed at 250 °C for 16 h.Powder XRD patterns were obtained on a Philips PW 1840 diffractometer equipped with a graphite monochromator using CuKα radiation. The powder XRD patterns at small angles were obtained with a Bruker D8 Discover X-ray diffractometer equipped with the 2-dimensional Hi-Star Area Detector and Cross Coupled Göbel Mirrors. The measurements were performed in transmission mode at room temperature using monochromatic CuKα1 radiation and a sample to detector face of 30 cm.
The elemental analysis of the materials was carried out using an ICP-OES 4300DV plasma emission spectrometer (Perkin Elmer, United States), enabling simultaneous recording of the full emission spectrum of the sample in the range from 166.25 to 847 nm with the help of a Charge Injection Device (CID). The spectrometer was equipped with a cyclonic spray chamber with concentric nebulizer. A double system of observation of plasma was used (axial and radial). Approximately 10–40 mg sample was weighed, and transported into a thoroughly rinsed Teflon vessel. All samples are dissolved in an aqua regia acid mixture containing 1.2 mL concentrated hydrochloric acid (36% v/v), 0.4 mL concentrated nitric acid (69% v/v) plus 0.4 mL concentrated hydrofluoric acid (40% v/v).
Temperature-programmed desorption (TPD) of ammonia was carried out on a Micromeritics TPR/TPD 2920 equipped with a thermal conductivity detector (TCD). The sample (30 mg) was pre-treated at 600 °C to remove volatile components. Prior to the TPD measurements the samples were degassed at 250 °C (after ramping from room temperature at a ramp rate of 10 °C min−1) for 1 h then saturated with ammonia gas at 100 °C for 30 min at a flow of 20 cm3 min−1. After a purge of ammonia by helium for 30 min at a flow of 10 cm3 min−1 at 100 °C, desorption of NH3 was monitored in the range between 100 and 600 °C at a ramp rate of 10 °C min−1.
The FT-IR spectra were measured on a Perkin Elmer Spectrum ONE instrument. A KBr wafer was prepared containing a particular amount of the calcined sample that allowed for a transmission of minimally 50% (<1 mg). The spectrum was taken over a range of 450–4000 cm−1 with a resolution of 1 cm−1.
Transmission electron microscopy was performed by using a Philips CM30T electron microscope with a LaB6 filament as source of electrons operated at 300 kV.
The MAS NMR was recorded on a Bruker Avance-400 Spectrometer with a 4 mm zirconium rotor sample holder spinning at 10 kHz. The 11B/27Al MAS NMR was measured at a frequency of 128.31/104.20 MHz with a spectral width of 25/100 kHz, acquisition time of 0.05 s/0.08 s, and acquisition delay 1 s, the total number of scans was 2000/4000. Liquid 11B NMR spectra were recorded with a Bruker Avance 400 spectrometer with chemical shifts (δ) reported in ppm downfield with B(OH)3 0.1 M in D2O.
HMF synthesis
Prins reaction
Acknowledgements
The authors gratefully acknowledge financial support from BeBasic and Dr Harald Ruijssenaars (Bird Engineering) for fruitful and valuable discussions about this work. The authors would like to acknowledge the TU Delft colleagues for their help in the different analyses: Willy Rook (physisorption), Dr Olav Steinebach (ICP-OES), Dr Patricia Kooyman (TEM), Ben Norder (XRD), Remco van Oosten (GC), Maarten Gorseling (HPLC, FTIR).Notes and references
- J. C. Jansen, Z. Shan, L. Marchese, W. Zhou, N. van der Puil and T. Maschmeyer, Chem. Commun., 2001, 713–714 RSC.
- M. S. Hamdy, PhD thesis, Delft University of Technology, 2005, open access on: http://repository.tudelft.nl/search/ir/?q=Hamdy&faculty=&department=&type=&year= Search PubMed.
- S. Telalovic, A. Ramanathan, G. Mul and U. Hanefeld, J. Mater. Chem., 2010, 20, 642–658 RSC.
- Z. Shan, E. Gianotti, J. C. Jansen, J. A. Peters, L. Marchese and T. Maschmeyer, Chem.–Eur. J., 2001, 7, 1437–1443 CrossRef CAS.
- C. Simons, U. Hanefeld, I. W. C. E. Arends, R. A. Sheldon and T. Maschmeyer, Chem.–Eur. J., 2004, 10, 5829–5835 CrossRef CAS PubMed.
- A. Ramanathan, M. S. Hamdy, U. Hanefeld and T. Maschmeyer, Catal. Lett., 2004, 95, 113–117 CrossRef.
- M. S. Hamdy, G. Mul, W. Wei, R. Anand, U. Hanefeld, J. C. Jansen and J. A. Moulijn, Catal. Today, 2005, 110, 264–271 CrossRef CAS.
- M. S. Hamdy, A. Ramanathan, T. Maschmeyer, U. Hanefeld and J. C. Jansen, Chem.–Eur. J., 2006, 12, 1782–1789 CrossRef CAS PubMed.
- A. Ramanathan, D. Klomp, J. A. Peters and U. Hanefeld, J. Mol. Catal. A: Chem., 2006, 260, 62–69 CrossRef CAS.
- A. Ramanathan, M. S. Hamdy, R. Parton, T. Maschmeyer, J. C. Jansen and U. Hanefeld, Appl. Catal., A, 2009, 355, 78–82 CrossRef CAS.
- S. Telalović, A. Ramanathan, J. F. Ng, R. Maheswari, C. Kwakernaak, F. Soulimani, H. C. Brouwer, G. K. Chuah, B. M. Weckhuysen and U. Hanefeld, Chem.–Eur. J., 2011, 17, 2077–2088 CrossRef PubMed.
- C. C. Aquino
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) , H. O. Pastore, A. F. Masters and T. Maschmeyer, ChemCatChem, 2011, 3, 1759–1762 CrossRef.
, H. O. Pastore, A. F. Masters and T. Maschmeyer, ChemCatChem, 2011, 3, 1759–1762 CrossRef. - G. Imran, M. P. Pachamuthu, R. Maheswari, A. Ramanathan and S. J. Sardhar Basha, J. Porous Mater., 2012, 19, 677–682 CrossRef CAS.
- L. Li, T. I. Koranyi, B. F. Sels and P. P. Pescarmona, Green Chem., 2012, 14, 1611–1619 RSC.
- M. M. Antunes, S. Lima, M. Pillinger and A. A. Valente, Molecules, 2012, 17, 3690–3707 CrossRef CAS PubMed.
- Z. Guo, C. Zhou, S. Hu, Y. Chen, X. Jia, R. Lau and Y. Yang, Appl. Catal., A, 2012, 419–420, 194–202 CrossRef CAS.
- S. Haddoum, I. Fechete, B. Donnio, F. Garin, D. Lutic and C. E. Chitour, Catal. Commun., 2012, 27, 141–147 CrossRef CAS.
- B. Wang, T. P. Ang and A. Borgna, Sci. Adv. Mater., 2011, 3, 1004–1010 CrossRef CAS.
- B. Wang, T. P. Ang and A. Borgna, Microporous Mesoporous Mater., 2012, 158, 99–107 CrossRef CAS.
- M. S. Hamdy and G. Mul, Catal. Sci. Technol., 2012, 2, 1894–1900 CAS.
- N. Gargiulo, F. Pepe and D. Caputo, J. Colloid Interface Sci., 2012, 367, 348–354 CrossRef CAS PubMed.
- S. Lima, M. M. Antunes, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Appl. Catal., A, 2010, 388, 141–148 CrossRef CAS.
- D. Liu, X.-Y. Quek, S. Hu, L. Li, H. M. Lim and Y. Yang, Catal. Today, 2009, 147, S51–S57 CrossRef CAS.
- X.-Y. Quek, D. Liu, W. N. E. Cheo, H. Wang, Y. Chen and Y. Yang, Appl. Catal., B, 2010, 95, 374–382 CrossRef CAS.
- K. Parkhomenko, A. Tyunyaev, L. M. M. Tejada, A. Dedov, A. Loktev, I. Moiseev and A. C. Roger, IOP Conf. Ser.: Mater. Sci. Eng., 2011, 19, 012008 CrossRef.
- C. Qi, J. Huang, S. Bao, H. Su, T. Akita and M. Haruta, J. Catal., 2011, 281, 12–20 CrossRef CAS.
- S. Mandal, A. SinhaMahapatra, B. Rakesh, R. Kumar, A. Panda and B. Chowdhury, Catal. Commun., 2011, 12, 734–738 CrossRef CAS.
- J. Zhou, Z. Hua, J. Shi, Q. He, L. Guo and M. Ruan, Chem.–Eur. J., 2009, 15, 12949–12954 CrossRef CAS PubMed.
- B. Karmakar, A. Sinhamahapatra, A. B. Panda, J. Banerji and B. Chowdhury, Appl. Catal., A, 2011, 392, 111–117 CrossRef CAS.
- L. Deloux and M. Srebnik, Chem. Rev., 1993, 93, 763–784 CrossRef CAS.
- T. Müller, K. Djanashvili, I. W. C. E. Arends, J. A. Peters and U. Hanefeld, Chem. Commun., 2013, 49, 361–363 RSC.
- T. A. Houston, B. L. Wilkinson and J. T. Blanchfield, Org. Lett., 2004, 6, 679–681 CrossRef CAS PubMed.
- N. Maraš and M. Kočevar, Helv. Chim. Acta, 2011, 94, 1860–1874 CrossRef.
- G. L. O'Connor and H. R. Nace, J. Am. Chem. Soc., 1955, 77, 1578–1581 CrossRef.
- J. ten Dam and U. Hanefeld, ChemSusChem, 2011, 4, 1017–1034 CrossRef CAS PubMed.
- J. ten Dam, F. Kapteijn, K. Djanashvili and U. Hanefeld, Catal. Commun., 2011, 13, 1–5 CrossRef CAS.
- T. S. Hansen, J. Mielby and A. Riisager, Green Chem., 2011, 13, 109–114 RSC.
- S. Kallus, J. Patarin, P. Caullet and A. C. Faust, Microporous Mater., 1997, 10, 181–188 CrossRef CAS.
- H. Koller, C. Fild and R. F. Lobo, Microporous Mesoporous Mater., 2005, 79, 215–224 CrossRef CAS.
- I. Lezcano-Gonzalez, A. Vidal-Moya, M. Boronat, T. Blasco and A. Corma, Phys. Chem. Chem. Phys., 2010, 12, 6396–6403 RSC.
- T. D. Conesa, J. M. Campelo, D. Luna, J. M. Marinas and A. A. Romero, Appl. Catal., B, 2007, 70, 567–576 CrossRef CAS.
- S. Liu, H. He, Z. Luan and J. Klinowski, J. Chem. Soc., Faraday Trans., 1996, 92, 2011–2015 RSC.
- I. Eswaramoorthi and A. K. Dalai, Microporous Mesoporous Mater., 2006, 93, 1–11 CrossRef CAS.
- T. D. Conesa, J. M. Hidalgo, R. Luque, J. M. Campelo and A. A. Romero, Appl. Catal., A, 2006, 299, 224–234 CrossRef CAS.
- R. de Ruiter, A. P. M. Kentgens, J. Grootendorst, J. C. Jansen and H. van Bekkum, Zeolites, 1993, 13, 128–138 CrossRef CAS.
- M. Adjdir, T. Ali-Dahmane and P. G. Weidler, C. R. Chim., 2009, 12, 793–800 CrossRef CAS.
- J. Zhang, M. Liu, C. Song and X. Guo, Microporous Mesoporous Mater., 2011, 139, 31–37 CrossRef CAS.
- A. Sayari, C. Danumah and I. L. Moudrakovski, Chem. Mater., 1995, 7, 813–815 CrossRef CAS.
- C. Fild, D. F. Shantz, R. F. Lobo and H. Koller, Phys. Chem. Chem. Phys., 2000, 2, 3091–3098 RSC.
- M. G. Dekamin, Z. Mokhtari and Z. Karimi, Sci. Iran., 2011, 18, 1356–1364 CrossRef CAS.
- J. K. Puri, R. Singh and V. K. Chahal, Chem. Soc. Rev., 2011, 40, 1791–1840 RSC.
- G. García, M. Falco, P. Crespo, S. Cabrera and U. Sedran, Catal. Today, 2011, 166, 60–66 CrossRef.
- S. Cabrera, J. El Haskouri, C. Guillem, J. Latorre, A. Beltrán-Porter, D. Beltrán-Porter, M. D. Marcos and P. Amorós, Solid State Sci., 2000, 2, 405–420 CrossRef CAS.
- A. Sonoda, N. Takagi, K. Ooi and T. Hirotsu, Bull. Chem. Soc. Jpn., 1998, 71, 161–166 CrossRef CAS.
- M. Bonzcek and H. Follner, Monatsh. Chem., 1976, 107, 283–288 CrossRef CAS.
- D. J. Kim, Y. Hong, S. H. Kim, K. M. Lee, S.-d. Mun, S. Yoon, J. Lee, Y. Do and Y. Kim, Inorg. Chim. Acta, 2011, 378, 311–314 CrossRef CAS.
- B. Samran, T. J. White and S. Wongkasemjit, J. Porous Mater., 2011, 18, 167–175 CrossRef CAS.
- A. Ramanathan, M. C. Castro Villalobos, C. Kwakernaak, S. Telalovic and U. Hanefeld, Chem.–Eur. J., 2008, 14, 961–972 CrossRef CAS PubMed.
- A. Ramanathan, R. Maheswari and U. Hanefeld, J. Catal., 2006, 242, 82–91 CrossRef.
- S. Lima, M. M. Antunes, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Molecules, 2010, 15, 3863–3877 CrossRef CAS PubMed.
- S. Telalović, S. K. Karmee, A. Ramanathan and U. Hanefeld, J. Mol. Catal. A: Chem., 2013, 368–369, 88–94 CrossRef.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–794 RSC.
- Y. B. Yi, J. L. Lee, Y. H. Choi, S. M. Park and C. H. Chung, Environ. Chem. Lett., 2012, 10, 13–19 CrossRef CAS.
- Y. J. Pagan-Torres, T. Wang, J. M. Gallo, B. H. Shanks and J. A. Dumesic, ACS Catal., 2012, 2, 930–934 CrossRef CAS.
- H. Jadhav, E. Taarning, C. M. Pedersen and M. Bols, Tetrahedron Lett., 2012, 53, 983–985 CrossRef CAS.
- X. Guo, Q. Cao, Y. Jiang, J. Guan, X. Wang and X. Mu, Carbohydr. Res., 2012, 351, 35–41 CrossRef CAS PubMed.
- A. J. Crisci, M. H. Tucker, M.-Y. Lee, S. G. Jang, J. A. Dumesic and S. L. Scott, ACS Catal., 2011, 1, 719–728 CrossRef CAS.
- V. Degirmenci, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, ChemCatChem, 2011, 3, 969–972 CrossRef CAS.
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257–268 CrossRef CAS.
- V. V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijhuis, J. Catal., 2012, 287, 68–75 CrossRef CAS.
- T. Ståhlberg, S. Rodriguez-Rodriguez, P. Fristrup and A. Riisager, Chem.–Eur. J., 2011, 17, 1456–1464 CrossRef PubMed.
- H. J. Prins, Chem. Weekbl., 1917, 14, 627–630 CAS.
- H. J. Prins, Chem. Weekbl., 1917, 14, 932–939 CAS.
- H. J. Prins, Chem. Weekbl., 1919, 16, 1072–1073 CAS.
- I. M. Pastor and M. Yus, Curr. Org. Chem., 2007, 11, 925–957 CrossRef CAS.
- I. M. Pastor and M. Yus, Curr. Org. Chem., 2012, 16, 1277–1312 CrossRef CAS.
- R. Noyori, Adv. Synth. Catal., 2003, 345, 15–32 CrossRef CAS.
- Y. Nakatani and K. Kawashima, Synthesis, 1978, 147–148 CrossRef CAS.
- M. Fuentes, J. Magraner, C. De Las Pozas, R. Roque-Malherbe, J. P. Pariente and A. Corma, Appl. Catal., 1989, 47, 367–374 CrossRef CAS.
- A. Corma and M. Renz, Chem. Commun., 2004, 550–551 RSC.
- F. Iosif, S. Coman, V. Parvulescu, P. Grange, S. Delsarte, D. D. Vos and P. Jacobs, Chem. Commun., 2004, 1292–1293 RSC.
- D.-L. Shieh, C.-C. Tsai and A.-N. Ko, React. Kinet. Catal. Lett., 2003, 79, 381–389 CrossRef CAS.
- Z. Yongzhong, N. Yuntong, S. Jaenicke and G.-K. Chuah, J. Catal., 2005, 229, 404–413 CrossRef.
- Y. Nie, G.-K. Chuah and S. Jaenicke, Chem. Commun., 2006, 790–792 RSC.
- P. Mertens, F. Verpoort, A.-N. Parvulescu and D. De Vos, J. Catal., 2006, 243, 7–13 CrossRef CAS.
- A. F. Trasarti, A. J. Marchi and C. R. Apesteguía, J. Catal., 2007, 247, 155–165 CrossRef CAS.
- Y. Nie, S. Jaenicke and G.-K. Chuah, Chem.–Eur. J., 2009, 15, 1991–1999 CrossRef CAS PubMed.
- F. Neaţu, S. Coman, V. Pârvulescu, G. Poncelet, D. De Vos and P. Jacobs, Top. Catal., 2009, 52, 1292–1300 CrossRef.
- F. Vermoortele, M. Vandichel, B. Van de Voorde, R. Ameloot, M. Waroquier, V. Van Speybroeck and D. E. De Vos, Angew. Chem., Int. Ed., 2012, 51, 4887–4890 CrossRef CAS PubMed.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485–493 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra44406f |
| This journal is © The Royal Society of Chemistry 2013 |