DOI:
10.1039/C3RA44192J
(Paper)
RSC Adv., 2013,
3, 20647-20654
Bis(4′-(4-pyridyl)-2,2′:6′,2′′-terpyridine)ruthenium(II) complexes and their N-alkylated derivatives in catalytic light-driven water oxidation
Received 24th April 2013, Accepted 19th August 2013
First published on 11th September 2013
Abstract
Hydrogen sulfate salts of [Ru(1)2]2+ where 1 = 4′-(4-pyridyl)-2,2′:6′,2′′-terpyridine and four N-alkylated derivatives [Ru(L)2]4+ were used as photosensitizers (λmax ∼510 nm) for water oxidation in light driven reactions with peroxydisulfate as a sacrificial electron acceptor and Na10[Co4(H2O)2(α-PW9O34)2] (Co4POM) as the catalyst in sodium borate buffers at pH 8.0 and 9.0. The N-substituents investigated were benzyl (L+ = 2+), ethyl (L+ = 3+), allyl (L+ = 4+) and 4-cyanobenzyl (L+ = 5+). The O2 yield in the presence of [Ru(L)2]4+ (L+ = 2+–4+) was comparable to that obtained in the presence [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) using light sources with λmax ≈ 490 nm. The ruthenium(III) complexes [Ru(1)2]3+ and [Ru(L)2]5+ (L+ = 2+–5+) are rather unstable in acidic conditions and could not be isolated. The most efficient photosensitizers [Ru(L)2]5+ (L+ = 2+ and 4+) were the least stable under weakly basic conditions (pH 9.0) with a half-life τ1/2 ∼ 10 ms. The stability of the complexes under photocatalytic turnover conditions is probably controlled by the rate at which ligand L+ is oxidized by Co4POM in its highest oxidation state.
Introduction
The world's fossil energy resources are rapidly diminishing and the search for renewable and sustainable sources to replace these is becoming paramount. It has become clear that water splitting to dihydrogen and dioxygen by artificial photosynthesis reactions would be an ideal way to convert solar energy into a renewable fuel.1,2 This reaction is thermodynamically unfavorable by 1.23 V and requires the input of four photons with λ < 1000 nm.3–6 Overall, water oxidation is a 4-electron process, but the first step, 1-electron oxidation to hydroxyl radical, is prohibitively unfavorable, E = 2.85 V (vs. NHE at pH 0). A highly efficient water oxidation catalyst (WOC) is required to form O2 at a reasonable rate with low overpotential. Significant progress in the development of WOCs has been reported recently, and an all inorganic catalyst based on earth abundant Co-atoms embedded in a polyoxometalate framework, Na10[Co4(H2O)2(α-PW9O34)2] (Co4POM), has been described.7–9 For homogeneous light-driven water oxidation, a three-component system composed of a photosensitizer, a sacrificial electron acceptor and the WOC is generally used.10–13 Salts of [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) are the most commonly used sensitizers because of an intense absorption band at 450 nm (ε = 1.42 × 104 M−1 cm−1) and high oxidation potentials (E = 1.26 V vs. NHE). The photophysical and photochemical properties of [Ru(bpy)3]2+ derivatives have been a research focus for three decades,14–17 and are central to photo-driven electron transfer in dye sensitized solar cells (DSSCs).18–21 Recently two groups of researchers raised the question as to whether Co4POM is stable and acts as a homogeneous WOC or, rather, functions as a precursor to cobalt oxide or a different soluble species that is the actual WOC. These groups used quite different reaction conditions, including far higher Co4POM concentrations, and conducted different experiments than we did in our original7 work. Specifically, Stracke and Finke22 showed that under electrocatalytic conditions and 0.5 mM Co4POM, a cobalt oxide film forms on the electrode surface and is the dominant WOC under these conditions. However, this group recently reported that at the Co4POM concentrations used in our original work,7 soluble Co4POM could be a dominant catalyst,23 as all our experiments then and since have shown it to be.24 The second group demonstrated enhanced activity for first electron removal from Co4POM by photogenerated [Ru(bpy)3]3+ oxidant with aging time of Co4POM in solution. However, this is not water oxidation (a four-electron-four-proton process) and these investigators did not monitor O2 or study water oxidation.24,25 They also found no formation of cobalt oxide from Co4POM under their homogeneous conditions. Thus, this work, while reporting an interesting finding, is only marginally relevant to the other studies by our group, Stracke–Finke, and others. Other groups have since confirmed that Co4POM functions as a homogeneous catalyst under a range of conditions, or as a precursor to heterogeneous cobalt oxide under other conditions.26In this work we explore the use of [Ru(L)2]4+ complexes which are N,N′-dialkylated derivatives of bis(4′-(4-pyridyl)-2,2′:6′,2′′-terpyridine)ruthenium(II) as photosensitizers for water oxidation with long wavelength light. The N-substituents investigated are benzyl (L+ = 2+), ethyl (L+ = 3+), allyl (L+ = 4+) and 4-cyanobenzyl (L+ = 5+), Scheme 1. Co4POM was used as a typical WOC to evaluate the efficiency of this family of photosensitizers as it has been comprehensively characterized in solution7–9 and has a high WOC activity in light-driven conditions with prototype [Ru(bpy)3]2+ photosensitizers.
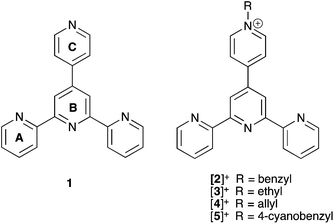 |
| | Scheme 1 Structures of ligands, and ring labelling in 1 for spectroscopic assignments. | |
Experimental section
General
[Ru(bpy)3]Cl2·H2O, sodium peroxydisulfate and all other purchased chemicals were of the highest purity available from commercial sources. [Ru(bpy)3]Cl2·H2O was recrystallized before use8,10 and [Ru(bpy)3][ClO4]3 was prepared as previously reported.10,27 The borate buffer was prepared by mixing 0.16 M (based on B) Na2B4O7 and H3BO3 solutions to achieve the desired pH. The compounds [Ru(2)2][HSO4]4, [Ru(3)2][HSO4]4, [Ru(4)2][HSO4]4 and [Ru(5)2][HSO4]4 were prepared as previously reported.28Electronic absorption and emission spectra were recorded using an Agilent 8453 spectrophotometer and a Shimadzu RF-5301 PC spectrofluorometer, respectively. Lifetime measurements were made using a Mini-Tau spectrometer from Edinburgh Instruments (475 nm laser diode) in air-equilibrated water. 1H and 13C NMR spectra were recorded on a Bruker DRX-500 MHz NMR spectrometer with 1H signals referenced to residual solvent peaks (TMS = δ 0 ppm); signals in the 13C NMR spectrum were referenced with respect to Na[Me3Si(CH2)3SO3] (DSS)29 with the SiMe3 signal at δ 0 ppm. Electrospray ionization mass spectra were recorded on a Bruker esquire 3000 plus mass spectrometer. Electrochemical data were obtained using (i) a BAS CV-50W electrochemical analyzer at room temperature equipped with a glassy-carbon working, a Pt-wire auxiliary, and a Ag/AgCl (3 M NaCl) BAS reference electrodes; or (ii) an Eco Chemie Autolab PGSTAT 20 system with glassy carbon working and platinum auxiliary electrodes. All redox potentials are reported relative to Ag/AgCl (3 M NaCl) reference electrode.
The fast reactions were studied using a Hi-Tech KinetAsyst Stopped Flow SF-61SX2 instrument equipped with a diode array detector operating in wavelength range 400–700 nm. Detailed analysis of kinetic data was performed using both Copasi 4.7 (Build 34)30 and the Solver subprogram in Microsoft Excel.
Light-induced water oxidation was carried out in the cylindrical quartz optical cell (NSG, 32G10) with a 1 cm optical path length, an outer diameter 22 mm, and total volume ∼2.8 mL equipped with standard joint. In a typical reaction, the vessel was filled with 2 mL of a solution with the desired concentrations of [Ru(bpy)3]Cl2·H2O, Na2S2O8, catalyst, and sodium borate buffer. The vessel was then sealed with a rubber septum and purged with Ar. The headspace was checked by gas chromatography (GC) to confirm the absence of O2 before the experiment. All procedures were performed with a minimum exposure to ambient light. The reaction was initiated by turning on the LED light source (LLS) equipped with a 490 nm LED. A light beam with a diameter 0.4–0.5 cm and 7 mW power was focused on the flat front of the reaction vessel using two lenses. The power of the light source was measured using a laser power meter Molectron, model Max 500A. The solution was agitated by a magnetic stirring bar spinning vertically on the back side of the cell. After the desired illumination time, the reaction was temporarily stopped by blocking the light. The O2-yield was quantified by GC as described earlier.8
[Ru(1)2][HSO4]2
[Ru(1)2][PF6]2 (150 mg, 0.164 mmol) and [nBu4N][HSO4] (300 mg, 0.884 mmol) were added to a mixture of MeCN and CH2Cl2 (6 mL, 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume) and the solution stirred for 30 min. A red precipitate formed and was separated by filtration. The solid was washed with Et2O (30 mL) and [Ru(1)2][HSO4]2 was isolated as a red solid (107 mg, 78.7%). 1H NMR (500 MHz, D2O) δ/ppm 9.30 (s, 4H, HB3), 9.13 (d, J = 6.6 Hz, 4H, HC2), 8.83 (d, J = 6.7 Hz, 4H, HC3), 8.72 (d, J = 8.1 Hz, 4H, HA3), 7.99 (td, J = 8.0, 1.2 Hz, 4H, HA4), 7.46 (d, J = 5.4 Hz, 4H, HA6), 7.22 (m, 4H, HA5). 13C NMR (126 MHz, D2O) δ/ppm 160.1 (CA2/CB2), 158.8 (CA2/CB2), 156.9 (CC4), 154.9 (CA6), 145.2 (CC2), 144.5 (CB4), 141.2 (CA4), 130.5 (CA5), 128.4 (CC3), 127.6 (CA3), 124.7 (CB3). ESI-MS m/z 361.0 [M − 2HSO4]2+ (base peak, calc. 361.1). UV-vis (H2O, 1.2 × 10−6 mol dm−3) λmax/nm (εmax/dm3 mol−1 cm−1) 490 (33100), 313 (58100), 274 (75500), 239 (39600). Emission (H2O, λexc = 490 nm) λem = 660 nm. Found C, 48.07; H, 3.63; N, 11.29; C40H30N8O8RuS2·4.5H2O requires C, 48.19; H, 3.94; N, 11.24 (see text).
:1 by volume) and the solution stirred for 30 min. A red precipitate formed and was separated by filtration. The solid was washed with Et2O (30 mL) and [Ru(1)2][HSO4]2 was isolated as a red solid (107 mg, 78.7%). 1H NMR (500 MHz, D2O) δ/ppm 9.30 (s, 4H, HB3), 9.13 (d, J = 6.6 Hz, 4H, HC2), 8.83 (d, J = 6.7 Hz, 4H, HC3), 8.72 (d, J = 8.1 Hz, 4H, HA3), 7.99 (td, J = 8.0, 1.2 Hz, 4H, HA4), 7.46 (d, J = 5.4 Hz, 4H, HA6), 7.22 (m, 4H, HA5). 13C NMR (126 MHz, D2O) δ/ppm 160.1 (CA2/CB2), 158.8 (CA2/CB2), 156.9 (CC4), 154.9 (CA6), 145.2 (CC2), 144.5 (CB4), 141.2 (CA4), 130.5 (CA5), 128.4 (CC3), 127.6 (CA3), 124.7 (CB3). ESI-MS m/z 361.0 [M − 2HSO4]2+ (base peak, calc. 361.1). UV-vis (H2O, 1.2 × 10−6 mol dm−3) λmax/nm (εmax/dm3 mol−1 cm−1) 490 (33100), 313 (58100), 274 (75500), 239 (39600). Emission (H2O, λexc = 490 nm) λem = 660 nm. Found C, 48.07; H, 3.63; N, 11.29; C40H30N8O8RuS2·4.5H2O requires C, 48.19; H, 3.94; N, 11.24 (see text).Results and discussion
Synthesis and characterization of [Ru(1)2][HSO4]2
The water-soluble complex [Ru(1)2][HSO4]2 was prepared via anion exchange by treating [Ru(1)2][PF6]231,32 with [nBu4N][HSO4].28 The red complex [Ru(1)2][HSO4]2 is insoluble in most common organic solvents but readily soluble in water. The electrospray mass spectrum exhibited a peak at m/z 361.0 assigned to the [M − 2HSO4]2+ ion. 1H and 13C NMR spectra were recorded in D2O and the latter were referenced to DSS (see Experimental section). The spectra were assigned by use of COSY, DEPT, HMQC and HMBC methods and were consistent with a single ligand environment in the [Ru(1)2]2+ ion. Elemental analysis of the complex indicated the formation of the hydrate [Ru(1)2][HSO4]2·4.5H2O. This was not unexpected in the light of data reported by us for a series of related complexes including [Ru(2)2][HSO4]4, [Ru(3)2][HSO4]4, [Ru(4)2][HSO4]4 and [Ru(5)2][HSO4]4.28 The electronic absorption spectrum of an aqueous solution of [Ru(1)2][HSO4]2 is similar to that of [Ru(1)2][PF6]2.31 Three high-energy absorptions at 313, 274 and 239 nm are assigned to ligand-based π* ← π transitions, and the band at 490 nm responsible for the red colour of [Ru(1)2][HSO4]2 arises from an MLCT transition. Excitation of [Ru(1)2][HSO4]2 at 490 nm results in an emission at 690 nm with a lifetime of 73 ns. This is somewhat shorter than the emission lifetimes observed for [Ru(L)2][HSO4]4 (L+ = 2+, 3+, 4+ and 5+). The quantum yield of 0.0018 for [Ru(1)2][HSO4]2 is of the same order of magnitude as those observed for [Ru(L)2][HSO4]4 (L+ = 2+, 3+, 4+ and 5+).28 The UV-vis spectroscopic and electrochemical data of [Ru(1)2][HSO4]2 were compared with those of [Ru(L)2][HSO4]4 (L+ = 2+–5+) complexes in water and are summarized in Table 1.
Table 1 UV-vis spectroscopic and electrochemical data for ruthenium(II) complexes in water
| Complex | λmax/nm | ε/dm3 mol−1 cm−1 | Ea/V | ΔEb/V | τem/ns |
|---|
| In the presence of 0.1 M NaHSO4 electrolyte, vs. Ag/AgCl reference electrode. Difference in potentials between [Ru(L)2](n+1)+/[Ru(L)2]n+ (L = 1, n = 2; L = 2–5, n = 4) and/[Ru(bpy)3]3+/[Ru(bpy)3]2+ couples. Ref. 33. |
|---|
| [Ru(bpy)3]Cl2 | 450 | 14000 | 1.028 | 0 | 550c |
| [Ru(1)2][HSO4]2 | 490 | 33000 | 1.21 | 0.182 | 73 |
| [Ru(2)2][HSO4]4 | 511 | 38000 | 1.24 | 0.212 | 135 |
| [Ru(3)2][HSO4]4 | 508 | 25000 | 1.21 | 0.192 | 146 |
| [Ru(4)2][HSO4]4 | 511 | 32000 | 1.21 | 0.182 | 137 |
| [Ru(5)2][HSO4]4 | 513 | 34000 | 1.24 | 0.212 | 108 |
Mechanistic evaluation of catalytic activity
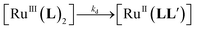
Throughout the mechanistic discussions, we use the following abbreviated formulae for clarity: [RuII(L)2] stands for [Ru(L)2]n+ (L = 1, n = 2; L+ = 2+–5+, n = 4); [RuIII(L)2] stands for [Ru(L)2](n+1)+ (L = 1, n + 1 = 3; L+ = 2+–5+, n + 1 = 5); [RuII(bpy)3] stands for [Ru(bpy)3]2+, and [RuIII(bpy)3] stands for [Ru(bpy)3]3+.The most common approaches for evaluating catalytic water oxidation activity in homogeneous systems are based on the use of strong oxidants such as Ce(IV) (Eo = 1.72 V vs. NHE34) or [Ru(bpy)3]3+ (Eo = 1.26 V vs. NHE16), or light induced generation of a strong oxidant such as [Ru(bpy)3]3+ in the presence of a sacrificial electron acceptor (often S2O82−). In the presence of light, the [RuII(L)2] or [RuII(bpy)3] driven water oxidation involves two key processes: (i) photoinduced oxidation of [RuII(L)2] to [RuIII(L)2] (or [RuII(bpy)3] to [RuIII(bpy)3]) by Na2S2O8; (ii) four sequential one-electron oxidation dark reactions of the catalyst to form O2-releasing species by [RuIII(L)2] or [RuIII(bpy)3]. This first photoinduced electron-transfer has been thoroughly studied for the reaction between [Ru(mptpy)2]4+ (mptpy+ = 4′-(4-methylpyridinio)-2,2′:6′,2′′-terpyridine) and Na2S2O8.35 The second process can be studied directly by observing the kinetics of [RuIII(L)2] reduction in water in the presence of a catalyst. Attempts to synthesize [RuIII(L)2] (L = 1 or L+ = 2+ or 4+) by oxidation with PbO2 in 0.5–1.0 M aqueous H2SO4 in a manner analogous to the synthesis of [RuIII(bpy)3] were unsuccessful due to the instability of the higher oxidation states. After addition of PbO2, the solution became dark green, but when the PbO2 powder was filtered off, the solution again became red (i.e. the colour characteristic of [RuII(L)2]).
To estimate the lifetime of [RuIII(L)2] (L = 1 or L+ = 2+ or 4+) in acidic conditions (0.5 M H2SO4), we generated the compounds using Ce(NH4)4(SO4)4·2H2O as a stoichiometric oxidant, eqn (1).
| | | [RuII(L)2] + Ce(IV) → [RuIII(L)2] + Ce(III) | (1) |
The reaction kinetics were followed by the formation of [RuIII(L)2] as monitored by an increase in an absorbance at 670 nm. One of the feeding syringes was filled with aqueous [RuII(L)2] and the second with Ce(NH4)4(SO4)4·2H2O in 0.5 M H2SO4. The concentration of [RuII(L)2] in the stock solutions was quantified using the absorbance at 510 nm. An example of the changes in absorbance is given in Fig. 1.
![Kinetics of formation and decomposition of [RuIII(2)2] (inset, longer time scale) in the reaction of 0.85 mM [RuII(2)2] with 0.62 mM Ce(NH4)4(SO4)4·2H2O in 0.5 M H2SO4 (red line) following the change in absorbance at 670 nm. The fitting to eqn (12) with k12 = 2.5 × 108 M−1 s−1 and ε4(670) = 430 M−1 cm−1 is shown by the dashed black line.](/image/article/2013/RA/c3ra44192j/c3ra44192j-f1.gif) |
| | Fig. 1 Kinetics of formation and decomposition of [RuIII(2)2] (inset, longer time scale) in the reaction of 0.85 mM [RuII(2)2] with 0.62 mM Ce(NH4)4(SO4)4·2H2O in 0.5 M H2SO4 (red line) following the change in absorbance at 670 nm. The fitting to eqn (12) with k12 = 2.5 × 108 M−1 s−1 and ε4(670) = 430 M−1 cm−1 is shown by the dashed black line. | |
The half-life of [RuIII(L)2] was estimated from the slow decrease of absorbance at 670 nm, A(670), to be ∼0.5 h. This is much shorter than for [RuIII(bpy)3]. This decomposition is thought to proceed via the reactions in eqn (2) and (3), where L′ is a one-electron oxidized ligand and P is the two-electron oxidized [RuII(L)2]. The estimated rate constants kd are in the range (0.5–5) × 10−3 s−1 and are given in Table 2.
| |  | (2) |
| | | [RuIII(L)2] + [RuII(L)(L′)] → P + [RuII(L)2] fast | (3) |
Table 2 The reaction rate constants of [RuIII(L)2] self-decomposition in 80 mM sodium borate buffer compared to that of [RuIII(bpy)3]
| Complex | pH 8 | pH 9 | 0.5 M H2SO4 |
|---|
| In 80 mM sodium phosphate buffer at pH 8. These complexes have a very weak effect on the rate of [RuIII(bpy)3] decomposition. Ref. 27 and 37. |
|---|
| [RuIII(bpy)3] | 0.012 | 0.05 | <5 × 10−5c |
| [RuIII(1)2] | <5b | n.d. | 5 × 10−4 |
| [RuIII(2)2] | 95 ± 30 | 115 ± 30 | 5 × 10−3 |
| [RuIII(2)2] | <1a | | |
| [RuIII(4)2] | 20 ± 7 | 90 ± 30 | 5 × 10−4 |
| [RuIII(3)2] | <10b | <10b | n.d. |
| [RuIII(5)2] | <10b | n.d. | n.d. |
As noted earlier, the low stability of [RuIII(L)2] (L = 1 or L+ = 2+ or 4+) does not permit their isolation. However, their solution spectra could be recorded (Fig. 2). The solutions typically contain 1–3% of [RuII(L)2] and decomposition products, which strongly absorb light at λ < 550 nm. Therefore, spectra were measured in the range 550–950 nm. Compared to [RuIII(bpy)3], the absorbance maxima of [RuIII(L)2] are shifted by about 90 nm to longer wavelength and extinction coefficients are about 50% higher.
![Part of the visible absorption spectrum of [RuIII(1)2] (green), [RuIII(2)2] (blue) and [RuIII(4)2] (red) in 0.5 M aqueous H2SO4.](/image/article/2013/RA/c3ra44192j/c3ra44192j-f2.gif) |
| | Fig. 2 Part of the visible absorption spectrum of [RuIII(1)2] (green), [RuIII(2)2] (blue) and [RuIII(4)2] (red) in 0.5 M aqueous H2SO4. | |
These experiments clearly indicate that [RuIII(L)2] is unstable even at high acidity. The low stability and short life-time of [RuIII(L)2] excited states could be prohibitive for use of these complexes as photosensitizers in light driven water oxidation reaction, although their higher oxidation potentials are advantageous.
To avoid precipitation of peroxydisulfate salts of [RuII(L)2], we have had to use relatively low concentrations in studies of the catalytic reactions. We have found that water is catalytically oxidized to O2 in the light driven system with S2O82− as a sacrificial electron acceptor in the presence of Co4POM (catalyst) and [RuII(L)2] (photosensitizer) in borate buffer at pH 8 and 9 (Fig. 3). The rates and O2 yields are strongly dependent on the ligand. Complexes of 1 and 5+ were almost inactive, those of 2+, 3+ and 4+ showed a similar initial activity, with [RuII(2)2] being the most efficient over a longer time scale. The benchmark photosensitizer [RuII(bpy)3] had a similar activity to [RuII(2)2]. The highest overall efficiency was obtained for [RuII(2)2] with O2 yields per peroxydisulfate (yield = 2 [O2]/[Na2S2O8]) of up to 30% and TON = (0.7–1.0) × 102. The yields were only weakly dependent on pH, although the rates were about double at pH 9. The turnover frequency, TOF, is commonly calculated as the ratio of the initial rate and the catalyst concentration, and [RuII(L)2] gave TOFs of up to ∼0.15 s−1 at pH 9. However, in light driven systems, the initial rate is actually proportional to the quantum yield but not related to the TOF.
![The time profile of O2 formation in light driven water oxidation reaction. Conditions: LED light source with λmax = 490 nm, 7 mW (photon flux 1.6 × 1016 photons s−1), total solution volume 2 mL, 2.5 mM Na2S2O8; 0.125 mM [RuII(L)2] or [RuII(bpy)3], 4 μM Co4POM, 80 mM borate buffer: (a) pH 8.0, and (b) at pH 9.0. Blue curve is for [RuII(bpy)3], black for [RuII(2)2], red for [RuII(4)2], green for [RuII(3)2], purple for [RuII(1)2] and dark red for [RuII(5)2].](/image/article/2013/RA/c3ra44192j/c3ra44192j-f3.gif) |
| | Fig. 3 The time profile of O2 formation in light driven water oxidation reaction. Conditions: LED light source with λmax = 490 nm, 7 mW (photon flux 1.6 × 1016 photons s−1), total solution volume 2 mL, 2.5 mM Na2S2O8; 0.125 mM [RuII(L)2] or [RuII(bpy)3], 4 μM Co4POM, 80 mM borate buffer: (a) pH 8.0, and (b) at pH 9.0. Blue curve is for [RuII(bpy)3], black for [RuII(2)2], red for [RuII(4)2], green for [RuII(3)2], purple for [RuII(1)2] and dark red for [RuII(5)2]. | |
At constant light intensity or constant photon flux (qp) and at high solution optical density, all light is absorbed and the total number of consumed photons during the time dt is dN = qpdt. The apparent (incident) quantum yield Φap is given by d(O2)/dN = (1/qp)d(O2)/dt. For the LED light source with λmax = 490 nm and power 7 mW, the photon flux qp is ∼1.6 × 1016 photons per s. The initial Φap reaches up to 25% for [RuII(bpy)3] at pH 9. Of the new photosensitizers, [RuII(1)2] and [RuII(5)2] produce very low amounts of O2. The initial quantum yields Φap for [RuII(2)2], [RuII(3)2] and [RuII(4)2] are similar and fall in the range 3–6% and 8–12% at pH 8 and 9, respectively. However, the reaction practically stops after about 10 minutes for [RuII(3)2] and [RuII(4)2], but continues longer for [RuII(2)2]. Based on [RuII(L)2] (L = 1 or L+ = 2+–5+), the turnover number TONRu = 4[O2]/[RuII(L)2], is about 10 for L = 2 at pH 9. There is no correlation of Φap and O2 yield with the oxidation potentials or excited state lifetimes of [RuII(L)2]. It is likely that the performance of the dyes is related to their stability under turnover conditions. Hypothetically, the amount of degraded dye can be determined from the difference in solution absorbance before and after the reaction. However, the products of dye oxidation also absorb at similar wavelengths. In addition, decomposition of one molecule of dye may require large numbers of oxidative equivalents. As a result, UV-vis spectroscopy becomes uninformative. As seen from Fig. 4, the absorbance after reaction for the most efficient dye [RuII(2)2] is smaller compared with much less efficient [RuII(1)2]. Oxidation of the ligand L by Ru(III) may proceed through: (i) an intramolecular pathway in [RuIII(L)2] (self-decomposition) in eqn (2) and (3); (ii) an oxidation of L by the catalyst in high oxidation state(s). The first pathway, eqn (2) and (3), is observed at neutral pH and is well known for [RuIII(bpy)3].
![The normalized absorption spectra of [RuII(3)2] (solid red line) and [RuII(1)2] (solid blue) before and after the light driven water oxidation reaction (dashed lines) at pH 9. Conditions are given in the caption to Fig. 3.](/image/article/2013/RA/c3ra44192j/c3ra44192j-f4.gif) |
| | Fig. 4 The normalized absorption spectra of [RuII(3)2] (solid red line) and [RuII(1)2] (solid blue) before and after the light driven water oxidation reaction (dashed lines) at pH 9. Conditions are given in the caption to Fig. 3. | |
In order to oxidize water, the reaction in eqn (2) should be slower than the rate of dioxygen formation kd[RuIII(L)2] ≪ −4d[O2]/dt. Since the rate of O2 formation at pH 9.0 is around 5 × 10−7 M−1s−1 and [RuIII(L)2] < 1.25 × 10−4 M, the self-decomposition might compete with water oxidation if kd > 10−3 s−1. The latter number is close to that determined in 0.5 M H2SO4. Because kd usually increases with pH, the reaction in eqn (2) could significantly decrease the O2 yield under turnover conditions at pH 8–9. The short life-times of [RuIII(L)2] do not allow their isolation and the direct measurements of their self-decomposition rate constants.
Rate constants of [RuIII(L)2] self-decomposition at elevated pH
As shown in Fig. 4, the concentration of ruthenium(II) species decreases in the course of the reaction as a result of self-decomposition of the photogenerated ruthenium(III) species. For all ligands, the rate constants of [RuIII(L)2] self-decomposition under turnover conditions were measured indirectly using the kinetic model in eqn (4)–(8). This model is based on an experimentally observed increase of the reaction rate (measured as dOD/dt, where OD is the optical density at 670 nm, A670) when a small amount of [RuIII(L)2] was added to [RuIII(bpy)3].| | | [RuIII(bpy)3] + [RuII(L)2] ⇌ [RuII(bpy)3] + [RuIII(L)2] | (5) |
| | | [RuIII(bpy)3] + P1 → [RuII(bpy)3] + P | (7) |
| | | [RuIII(bpy)3] + P2 → [RuII(bpy)3] + PP | (8) |
For simplicity we use abbreviations: [RuIII(bpy)3] = A, [RuII(L)2] = B, [RuII(bpy)3] = C, and [RuIII(L)2] = D. The D, P1 and P2 concentrations are considered as steady state. A thorough analysis of the data confirmed that the steady state concentration with respect to D is achieved in less than 1 ms. The reaction in eqn (5) is thermodynamically unfavourable: K5 = k5/k−5 is equal to 8.9 × 10−4 and 2.7 × 10−4 for ΔE = 0.182 and 0.212 V, respectively (Table 1). The electron transfer reactions between ruthenium polypyridine complexes, such as in eqn (5), are very fast and of the order of 109 M−1 s−1.36 Therefore under typical experimental conditions k−5C > 104 s−1 and it is reasonable to assume that k−5C ≫ k6. The mass balance in the reaction is as follows: A = A0 − D − P1, C = C0 + D, B = B0 − D; species P1 is a short lived intermediate and P1 ≪ D. After simplification, one obtains eqn (9).
| | | D = K5(A0 + D)(B0 − D)/(C0 − D) | (9) |
After simplification, eqn (9) gives eqn (10).
| | | D2 + [C0 + K5(A0 + B0)]D − K5A0B0 = 0 | (10) |
If:
then
eqn (10) becomes
eqn (11) and D is found from
eqn (12).
| | 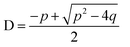 | (12) |
After reaching steady state conditions with respect to D, the decrease of optical density (OD) is described applying the Beer–Lambert law by eqn (13) (where l = path length). For simplicity, it was assumed that the molar extinction coefficients for P and PP are close to those of [RuII(bpy)3] and [RuII(L)2], respectively.
| | | d(OD)/dt = −d[(ε1A + ε2C + ε2P + ε3B + ε3PP + ε4D)l]/dt | (13) |
From the reaction mechanism in eqn (4)–(8), and after several standard mathematical transformations, the initial rate R of optical density decrease can be expressed as eqn (14).
| | | R = 2(ε1 − ε2)(k4A0 + k6D)l = R0 + 2(ε1 − ε2)k6Dl | (14) |
where
R0 = 2(
ε1 −
ε2)
k4A
0l is the initial rate of optical density decrease in the absence of [Ru
II(
L)
2] (the self-decomposition of [Ru
III(bpy)
3]).
Eqn (14) with D calculated from
eqn (12) was used to fit experimental data with
k6 as a variable parameter. Examples of the experimental and fitting data are presented in
Fig. 5, the values of
k6 are given in
Table 2. In the following section, we consider the influence of the structure of the ruthenium complexes on these rate constants and on the overall water photocatalytic oxidation reaction.
![Effect of [RuII(4)2] (a) and [RuII(bpy)3] (b) on initial rate of [RuIII(bpy)3] decomposition measured as the rate of decrease of absorbance at 670 nm, pH 9.0, 80 mM borate buffer. Initial concentrations: 0.45 mM [RuIII(bpy)3] (a and b), 5.610−5 M [RuII(bpy)3] (a), 2 × 10−6 M [RuII(bpy)3] (b), 0.08 mM [RuII(4)2] (b).](/image/article/2013/RA/c3ra44192j/c3ra44192j-f5.gif) |
| | Fig. 5 Effect of [RuII(4)2] (a) and [RuII(bpy)3] (b) on initial rate of [RuIII(bpy)3] decomposition measured as the rate of decrease of absorbance at 670 nm, pH 9.0, 80 mM borate buffer. Initial concentrations: 0.45 mM [RuIII(bpy)3] (a and b), 5.610−5 M [RuII(bpy)3] (a), 2 × 10−6 M [RuII(bpy)3] (b), 0.08 mM [RuII(4)2] (b). | |
Structure–activity correlation
As can be seen from Table 2, there is no simple correlation between the stability of the dyes (defined as the rate of self-decomposition, k6) and the potentials of the [RuIII(L)2]/[RuII(L)2] couples. The effect of pH is small: ruthenium(II) polypyridyl complexes are less stable with increasing pH (Table 2). Changing borate to phosphate buffer has a dramatic effect on the relative stabilities of the dyes. The self-decomposition of [RuIII(bpy)3] measured from the decrease of optical density at 670 nm obeys a simple exponential law at both pH 8 and 9. The reaction in phosphate buffer is faster but the kinetics are more complex.27 The addition of [RuII(2)2] to [RuIII(bpy)3] in phosphate buffer weakly affects the rate of [RuIII(bpy)3] decomposition. The formal application of eqn (14) gives k6 < 0.5 s−1, which is significantly lower than in borate buffer. Since the activity of Co4POM in water oxidation is much lower in phosphate than in borate buffer, the stabilities of the [RuIII(L)2] complexes were not studied in phosphate buffer.The dioxygen yield in a light-driven system is strongly dependent on the nature of the photosensitizer. Surprisingly, the most stable dye gave the lowest O2 yield. This indicates that an intramolecular oxidation of the ligand L is much slower than intermolecular oxidation by strong oxidants present in solution under turnover conditions. The O2 yield correlates to some extent with the lifetime of their excited states (Table 1). The quenching of the excited state [RuII(L)2]* by S2O82− proceeds through bimolecular and more efficient unimolecular pathways, eqn (15)–(17).
| | | [RuII(L)2] + S2O82− ⇌ {[RuII(L)2]⋯S2O82−} | (15) |
| | | [RuII(L)2]* + S2O82− → [RuIII(L)2] + SO42− + SO4˙− | (16) |
| | | {[RuII(L)2]⋯S2O82−}* → [RuIII(L)2] + SO42− + SO4˙− | (17) |
The lifetime of [RuII(L)2]* is in the range 73–146 ns and significantly shorter than that of [RuII(bpy)3]* (550 ns). The shorter lifetime results in lower quenching efficiency and lower quantum yield of O2 formation (ΦCY). In addition, the quenching efficiency for [RuII(1)2]* is lower since the {[RuII(L)2]⋯S2O82−} ion pair is less favourable. Consequently, the initial ΦCY for L = 2, 3 or 4 is higher than for L = 1. Since the self-decomposition is slower under turnover conditions, the stability of [RuIII(L)2] does not affect the final O2 yield. Interestingly, the reaction involving the most stable complex [RuIII(3)2] stops much earlier compared with that involving the least stable complex [RuIII(2)2]. The sulfate anion radical SO4˙− formed in eqn (16) and (17) is believed to selectively oxidize another [RuII(L)2] complex to [RuIII(L)2] (k = 5 × 109 M−1 s−1 for L = bpy).38 In our case this electron transfer pathway may compete with the reaction of SO4˙− with a ligand L. For example, SO4−˙ quickly reacts with allylic alcohol and benzene, ∼109 M−1 s−1, but slower with pyridine, ∼108 M−1 s−1.39 This would result in a faster degradation of [RuII(2)2] and [RuII(4)2] by SO4˙− than of [RuII(1)2]. However, this is not consistent with the data in Fig. 3. Such controversial behaviour is probably related to the oxidation of the ligand L by the water oxidation catalyst (WOC). In our system the WOC, namely Co4POM, is negatively charged and forms strong ion-pairs with [RuII(L)2] or [RuIII(L)2] (eqn (18)).40–42
After removal of four electrons from Co4POM, the reactive Co4POM(−4e) intermediate oxidizes water as in eqn (19). Being a strong oxidant, this intermediate may also oxidize the ligand L as shown in eqn (20).
| | | Ru(L)2 + Co4POM(-4e) ⇌ {Ru(L)2Co4POM(-4e)} | (18) |
| | | {Ru(L)2Co4POM(-4e)} + 2H2O → {Ru(L)2Co4POM} + O2 + 4H+ | (19) |
| | | {Ru(L)2Co4POM(-4e)} → {Ru(LL′)Co4POM} | (20) |
where
L′ is a product of ligand oxidation. As a result, the O
2 yield is dependent on the competition between the two processes in
eqn (19) and (20). The factors controlling oxidation of ligand
L by the catalyst are not well understood. Studies with different catalysts are in progress.
Conclusions
A family of N-alkylated derivatives of the complex [Ru(1)2]2+ (1 = 4′-(4-pyridyl)-2,2′:6′,2′′-terpyridine) has been investigated in conjunction with Co4POM for their catalytic water oxidation activity. These [Ru(L)2]4+ complexes (abbreviated “[RuII(L)2]”) have two properties which argue for their use as photosensitizers, namely the longer wavelength absorption and higher oxidation potential, compared to the current standard, [Ru(bpy)3]2+ (abbreviated “[RuII(bpy)3]”). In water oxidation experiments, complexes incorporating ligands 1 and 5+ were almost inactive with low O2 yields and a short reaction time. The activities of complexes incorporating ligands 2+, 3+ and 4+ were similar to one another, with [RuII(2)2] being the most efficient over a long time scale. [RuII(2)2] showed water oxidation activity that was comparable with the current standard photosensitizer, [RuII(bpy)3], with a TOF of up to 0.15 s−1, TON of 1 × 102 and a 30% O2 yield, based on the peroxydisulfate concentration. We propose that the performance of the dye is dependent on oxidizability of L by the catalyst in a high oxidation state. In order to confirm this, the rates of self-decomposition of [RuIII(L)2] were determined indirectly using a kinetic model. The dye containing ligand 3 is the most stable with respect to self-decomposition but gives the lowest O2 yield and the reaction stops much earlier compared with that involving the least stable complex (that with ligand 2). While the series of complexes is promising in terms of light absorption and higher oxidation potentials, further work needs to be carried out to develop a [RuII(L)2] photosensitiser which can generate higher O2 yields.Acknowledgements
We thank the U.S. National Science Foundation (grant number CHE-0911610), the Swiss National Science Foundation, the European Research Council (Advanced Grant 267816 LiLo) and the University of Basel for support of this research.Notes and references
- J. Chow, R. J. Kopp and P. R. Portney, Science, 2003, 302, 1528 CrossRef PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- R. Eisenberg and H. B. Gray, Inorg. Chem., 2008, 47, 1697 CrossRef CAS PubMed.
- T. A. Betley, Y. Surendranath, M. V. Childress, G. E. Alliger, R. Fu, C. C. Cummins and D. G. Nocera, Philos. Trans. R. Soc., B, 2008, 363, 1293 CrossRef CAS PubMed.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185 RSC.
- K. J. Young, L. A. Martini, R. L. Milot, R. C. Snoeberger III, V. S. Batista, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2012, 256, 2503 CrossRef CAS PubMed.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342 CrossRef CAS PubMed.
- Z. Huang, Z. Luo, Y. V. Geletii, J. W. Vickers, Q. Yin, D. Wu, Y. Hou, Y. Ding, J. Song, D. G. Musaev, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2011, 133, 2068 CrossRef CAS PubMed.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572 RSC.
- Y. V. Geletii, Z. Huang, Y. Hou, D. G. Musaev, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 7522 CrossRef CAS PubMed.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841 CrossRef CAS PubMed.
- C. Besson, Z. Huang, Y. V. Geletii, S. Lense, K. I. Hardcastle, D. G. Musaev, T. Lian, A. Proust and C. L. Hill, Chem. Commun., 2010, 2784 RSC.
- N. S. McCool, D. M. Robinson, J. E. Sheats and G. C. Dismukes, J. Am. Chem. Soc., 2011, 133, 11446 CrossRef CAS PubMed.
- W. J. Dressick, T. J. Meyer, B. Durham and D. P. Rillema, Inorg. Chem., 1982, 21, 3451 CrossRef CAS.
- B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4803 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. V. Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- E. H. Yonemoto, R. L. Riley, Y. I. Kim, S. J. Atherton, R. H. Schmehl and T. E. Mallouk, J. Am. Chem. Soc., 1992, 114, 8081 CrossRef CAS.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Müller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382 CrossRef CAS.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef PubMed.
- A. Delgadillo, M. Arias, A. M. Leiva, B. Loeb and G. J. Meyer, Inorg. Chem., 2006, 45, 5721 CrossRef CAS PubMed.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115 RSC.
- J. J. Stracke and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 14872 CrossRef CAS PubMed.
- J. J. Stracke and R. G. Finke, ACS Catal., 2013, 3, 1209 CrossRef CAS.
- J. W. Vickers, H. Lv, J. M. Sumliner, G. Zhu, Z. Luo, D. G. Musaev, Y. V. Geletii and C. L. Hill, J. Am. Chem. Soc, 2013, DOI:10.1021/ja4024868.
- M. Natali, S. Berardi, A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Commun., 2012, 48, 8808 RSC.
- S. Goberna-Ferrón, L. Vigara, J. Soriano-López and J. R. Galán-Mascarós, Inorg. Chem., 2012, 21, 11707 CrossRef PubMed.
- P. K. Ghosh, B. S. Brunschwig, M. Chou, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1984, 106, 4772 CrossRef CAS.
- E. C. Constable, M. Devereux, E. L. Dunphy, C. E. Housecroft, J. A. Rudd and J. A. Zampese, Dalton Trans., 2011, 40, 5505 RSC.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795 CrossRef CAS.
- S. Hoops, S. Sahle, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singhal, L. Xu, P. Mendes and U. Kummer, Bioinformatics, 2006, 22, 3067 CrossRef CAS PubMed.
- E. C. Constable and A. M. W. Cargill Thompson, Dalton Trans., 1994, 1409 RSC.
- E. C. Constable, E. L. Dunphy, C. E. Housecroft, W. Kylberg, M. Neuburger, S. Schaffner, E. R. Schofield and C. B. Smith, Chem.–Eur. J., 2006, 12, 4600 CrossRef CAS PubMed.
- H. S. White, W. G. Becker and A. J. Bard, J. Phys. Chem., 1984, 88, 1840 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, 2000 Search PubMed.
- A. L. Kaledin, Z. Huang, Q. Yin, E. L. Dunphy, E. C. Constable, C. E. Housecroft, Y. V. Geletii, T. Lian, C. L. Hill and D. G. Musaev, J. Phys. Chem. A, 2010, 114, 6284 CrossRef CAS PubMed.
- C. Creutz, M. Chou, T. L. Netzel, M. Okumura and N. Sutin, J. Am. Chem. Soc., 1980, 102, 1309 CrossRef CAS.
- B. S. Brunschwig, M. H. Chou, C. Creutz, P. Ghosh and N. Sutin, J. Am. Chem. Soc., 1983, 105, 4832 CrossRef CAS.
- K. Henbest, P. Douglas, M. S. Garley and A. Mills, J. Photochem. Photobiol., A, 1994, 80, 299 CrossRef CAS.
- P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 1027 CrossRef CAS.
- R. Ballardini, M. T. Gandolfi and V. Balzani, Inorg. Chem., 1987, 26, 862 CrossRef CAS.
- M. Natali, M. Orlandi, S. Berardi, S. Campagna, M. Bonchio, A. Sartorel and F. Scandola, Inorg. Chem., 2012, 51, 7324 CrossRef CAS PubMed.
- P.-E. Car, M. Guttentag, K. K. Baldridge, R. Alberto and G. R. Patzke, Green Chem., 2012, 14, 1680 RSC.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a