Multifaceted half-sandwich arene–ruthenium complexes: interactions with biomolecules, photoactivation, and multinuclearity approach
Sanjay Kumar
Singh
a and
Daya Shankar
Pandey
*b
aDiscipline of Chemistry, School of Basic Sciences, Indian Institute of Technology (IIT) Indore, Khandwa Road, Indore-452 017, Madhya Pradesh, India. E-mail: sksingh@iiti.ac.in; Fax: +91 731 2431 482; Tel: +91 731 2438 730
bDepartment of Chemistry, Faculty of Science, Banaras Hindu University, Varanasi-221 005, Uttar Pradesh, India. E-mail: dspbhu@bhu.ac.in; Fax: +91 542 2368 174; Tel: +91 542 7602 480
First published on 4th October 2013
Abstract
Biological properties of the arene–ruthenium complexes have attracted substantial current interest. Their activity is appreciably defined and controlled by the arene moieties, organic ligands and the metal center. In this review, we discuss the interaction of arene–ruthenium complexes with significant biomolecular targets (DNA and enzymes). Principally, active complexes may interact with the biomolecular targets DNA or nucleobases either by direct coordination facilitated by aquation of the complex or by intercalation/stacking of the pendant planar part of the complex, usually from the planar ancillary ligands or arenes with extended rings, between the DNA base pairs. On the other hand, kinetically inert metal complexes can also provide a potential tool (as enzyme inhibitors) for the targeting of important biomolecules (other than DNA), such as protein kinases. At the same time, coordination with a metal facilitates the outreach of the organic molecules in the intracellular region. This review also highlights the photodriven activation of arene–ruthenium complexes, important metal–drug interactions and the potential of multinuclear scaffolds as important drug candidates (e.g., metallodendrimers) and drug carriers (e.g. metallacages) for targeted delivery and activity.
 Sanjay Kumar Singh | Sanjay Kumar Singh received his PhD in Chemistry from A.P.S. University, Rewa, India in 2007. From 2008–2011 he was a JSPS postdoctoral fellow and then AIST postdoctoral scientist in the research group of Prof. Qiang Xu at AIST, Osaka, Japan. In 2011, he was an Alexander von Humboldt Postdoctoral Fellow in the research group of Prof. P.W. Roesky at Karlsruhe Institute of Technology, Karlsruhe, Germany. Currently, he is Assistant Professor of Chemistry at the Indian Institute of Technology (IIT) Indore, India. His research interests are the synthetic, organometallic and coordination chemistry of transition metals and nanoparticles for catalysis. |
 Daya Shankar Pandey | Daya Shankar Pandey received his PhD degree in Chemistry from the Indian Institute of Technology (IIT) Kanpur, India in 1989. He started his career as a lecturer in A.P.S. University, Rewa, India and currently, is a Professor in the Department of Chemistry at Banaras Hindu University (BHU), Varanasi, India. He is a Fellow of the National Academy of Sciences, India and is a recipient of the Chemical Research Society of India Bronze medal (2005). His research interests include piano-stool arene–ruthenium complexes based metallo-ligands and DNA cleaving agents. He has published research articles in various refereed journals. |
1. Introduction
Ruthenium complexes are unarguably among the most important potential candidates investigated as anticancer metallodrugs based on metals other than platinum.1–20 Their inherent properties like mimicking iron, redox accessible oxidation states, low toxicity, smooth interaction and binding with DNA and proteins, highlight their ability to act as metallodrugs. The ruthenium complexes containing imidazole and indazole are taken as classical models for anticancer activity. Among the numerous ruthenium complexes extensively studied, imidazolium trans-[tetrachloro(dimethylsulfoxide)imidazoleruthenium-(III)] (1, NAMI-A) and indazolium trans-[tetrachlorobis(1H-indazole)ruthenium(III)] (2, KP 1019), are currently under clinical trials (Fig. 1).21–24In vivo reduction of the Ru(III) prodrugs into activated Ru(II) complexes can be better explained by the “activation by reduction” hypothesis. Usually, tumor cells have a lower oxygen content (hypoxia) and lower pH (due to the excess production of lactic acid by glycolysis) relative to the surrounding normal cells, therefore a relatively high production of Ru(II) in tumor cells is favoured. Ru(II) complexes are more reactive and can coordinate biomolecules more firmly to provide selective toxicity.12,25–27 | ||
| Fig. 1 Important ruthenium based anticancer drug candidates. | ||
Among the ruthenium organometallic complexes, the half-sandwich arene–ruthenium complexes in particular have offered great promise for exploration as anticancer agents.1–20 This is due to their vast structural diversity and bonding modes e.g., the hydrophobic arene ligand facilitates diffusion of the drug through the cell membrane, whereas the three remaining coordination sites usually occupied by various mono-, bi- or tridentate ligands offer a diverse structural and coordination mode and modulate the biological properties of these complexes. In 1992, Tocher and co-workers presented arene–ruthenium complexes as anticancer agents, where an enhancement in the cytotoxicity was observed by introducing the anticancer agent metronidazole, [1-β-(hydroxyethyl)-2-methyl-5-nitroimidazole], to a benzene ruthenium dichloro fragment.28 Extensive investigation of the anticancer activities of arene–ruthenium complexes was pioneered by Sadler and Dyson.1,4,5,7,10,16,17,29–43 Arene–ruthenium(II) complexes [(η6-biphenyl)Ru(en)Cl][PF6] (3) (RM175, en = ethylenediamine), reported by Sadler and co-authors,44 and [(η6-p-cymene)Ru(pta)Cl2] (4) (RAPTA-C, pta = 1,3,5-triaza-7-phosphatricyclo[3.3.1.1]decane) developed by Dyson and co-workers (Fig. 2),45 have shown promising therapeutic potential and are considered to be prototypes of anticancer arene–ruthenium(II) complexes for the design and understanding of the mode of action of other anticancer drug candidates.
 | ||
| Fig. 2 Prototype arene–ruthenium complexes as anticancer agents. | ||
Despite extensive investigations in this direction, development and improvements in the selectivity and activity of such complexes is of prime importance to develop safe, selective and more effective arene–ruthenium based biological agents. Here we highlight the applications of biologically active arene–ruthenium(II) complexes in some areas of current interest. We focus especially on their interactions with biomolecular targets, including DNA and enzymes, photodriven activity, metal–drug interactions, and multinuclear scaffolds as drug candidates and drug delivery systems.
2. Interactions of arene–ruthenium complexes with DNA or nucleobases
DNA is considered to be one of the potential biomolecular targets for anticancer drugs. However, interaction of metallodrugs with DNA is not clearly understood. Arene–ruthenium complexes display different mechanisms for exhibiting anticancer activity. The acceptable mechanism involves initial aquation of the labile Ru–Cl bond to form intermediary Ru–aqua complexes, which further interact with DNA bases to form complex–DNA adducts. Apart from direct DNA binding, the arene–ruthenium complexes with extended arene, e.g. biphenyl, dihydroanthracene (dha), or the planar ancillary ligands may also intercalate between the DNA base pairs. In recent years, several reviews have appeared in the literature on the cytotoxicity of arene–ruthenium complexes.1,4–10,12,13,16,17 Here, we briefly summarise important and current potential arene–ruthenium complexes displaying interaction with DNA base pairs.Dyson and co-workers extensively evaluated the anticancer properties of the prototype arene–ruthenium(II) complex 4 (RAPTA-C) and its derivatives (Fig. 2 and 3).1,4,45–53 As RAPTA-type compounds possess a modular structure comprising a ruthenium-bound arene, one pta molecule and two halides, it is possible to design compounds potentially able to increase the drug activity.46 Although, 4 exhibits only a low activity in vitro, it is very active in vivo. Replacing the p-cymene with benzene (5), toluene (6) and hexamethylbenzene in RAPTA, the resulting complexes influence toxicity only slightly,1,47 whereas replacement of the arene by cyclopentadienyl (C5H5) in RAPTA, leads to the complex [(η6-C5H5)Ru(pta)2Cl] which is inactive for cancerous cells.48 However, replacement of the labile chloro groups with a chelating anionic ligand, oxalate (7) and diketonato (8), drastically enhances the stability of these complexes against easy hydrolysis and therefore, increases the impact on the cytotoxicity.38,49 Moreover, different enantiomers of RAPTA (9, 10), which have a chiral center on the arene ligand exhibit distinct differences in their activity. For instance, R-isomers are less cytotoxic than S-isomer against cancer cells. The observed differences in the activities are presumably due to specific interactions with relevant biomolecular targets, induced by the chiral centre of the complex.53 Mechanistic studies on the interaction of RAPTA complexes with DNA suggest that 4 preferentially binds to the purine base, guanine,51 and that the binding occurs by the loss of the chloro group and arene ligand, while pta remains coordinated.52
 | ||
| Fig. 3 Arene–ruthenium complexes with soluble phosphorous based ligand. | ||
Moreover, along with the potential anticancer activity, the arene–ruthenium complexes such as the RAPTA series of complexes with modified and structurally designed ligands (e.g. imidazole based ligands) also exhibit antimetastatic, antimalarial, antifilarial activity, and these effects are brought about by the synergistic combination of RAPTA complexes with the imidazole ligand, as observed for 1 (NAMI-A).11,54 Dyson and co-workers also investigated arene–ruthenium complexes containing imidazole such as [(η6-arene)Ru(imd-L)Cl2], [(η6-arene)Ru(imd-L)2Cl][X], and [(η6-arene)Ru(imd-L)3][X]2 (η6-arene = benzene, imd-L = N-methylimidazole (11); η6-arene = p-cymene, imd-L = N-vinylimidazole (12); X = Cl, BF4, BPh4) (Fig. 4).55 Though these exhibit lower cytotoxicity relative to cisplatin, their activity is comparable to the arene–ruthenium complexes of the RAPTA series. We also investigated the activity of arene–ruthenium complexes with imidazole based ligands [(η6-arene)Ru(CPI)Cl2] (CPI = 1-(4-cyanophenyl)-imidazole; η6-arene = benzene, p-cymene (13, RACPI), hexamethylbenzene) and analogous RAC derivatives [(η6-arene)Ru(CNPy)Cl2] (CNPy = 4-cyanopyridine) and [(η6-arene)Ru(L)(PPh3)Cl]+ (L = CPI (14) and CNPy) toward filarial parasite Setaria cervi.56 In contrast to the high potential of these complexes toward DNA binding, [(η6-arene)Ru(L)(CPI)]2+, (L = bipyridine, phenanthroline) are active Topo II inhibitors.56
 | ||
| Fig. 4 Arene–ruthenium complexes containing imidazole based ligands. | ||
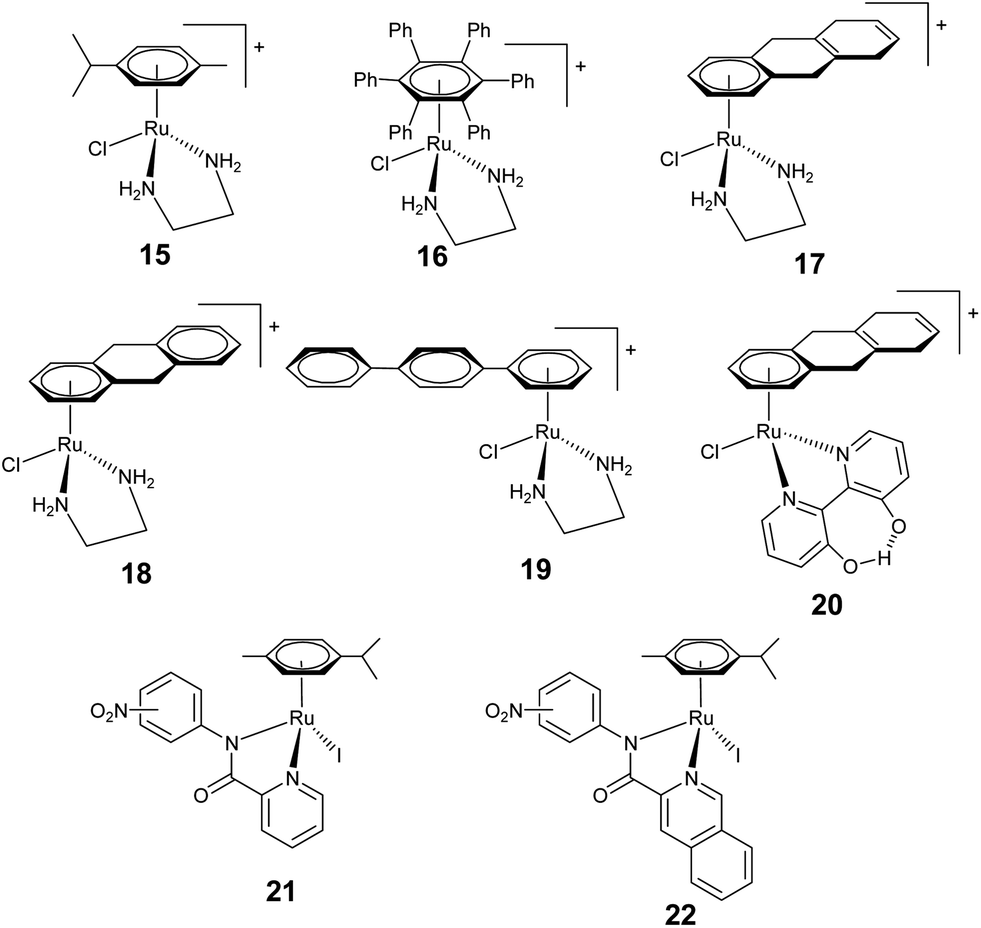
Sadler and co-workers reported a series of monofunctional [(η6-arene)Ru(en)X]+ complexes, having only one reactive Ru–Cl bond which represents one of the biologically most active class of complexes (Fig. 5).5,10,32,33,57–59 The Ru–Cl bond in these complexes undergoes rapid hydrolysis to give aqua complexes [(η6-arene)Ru(en)(H2O)]2+ that can bind to DNA or other biomolecules.60 In general, these complexes bind specifically to the nucleobase guanine through N7 in comparison to other nucleobases.44,57,61 The interaction of [(η6-arene)Ru(en)X]+ with DNA bases upon hydrolysis follows a two step process.62 The reaction first proceeded with the binding of the 5′-phosphate group which then underwent an intramolecular rearrangement to form the N7 purine–metal adduct. The activity of these complexes [(η6-arene)Ru(en)X]+ (η6-arene = p-cymene, 15; biphenyl, 3) is comparable to carboplatin,44 whereas with more hydrophobic arene, such as tetrahydroanthracene (tha, 17) the activity is equivalent to cisplatin.40 Moreover, the complexes bearing relatively non polar, sterically demanding alkyl, phenyl (16), or benzyl groups are more potent relative to the benzene analogue. Therefore, the arene unit has a significant effect on the cytotoxic behaviors of these complexes, particularly their size, flexibility and degree of hydrophobicity or lipophilicity. In general, the lipophilic character of the complex enhances its uptake into the cell whereas hydrophobic interactions between the arene and DNA bases facilitates the binding of the complex to DNA.63 The aquation process in this series of complexes increases with an increase in the size of arene ([(η6-dha)Ru(en)Cl]+ (18) > [(η6-tha)Ru(en)Cl]+ (17) > [(η6-biphenyl)Ru(en)Cl]+ (3)). Further extension of the additional phenyl ring, e.g. in [(η6-p-terphenyl)Ru(en)Cl]+ (19) with o- and m-terphenyl arene results in a much higher activity and even comparable potency to cisplatin. Upon aquation the complex binds to DNA rapidly, specifically to N7 of the guanine and causes significant DNA unwinding.35
 | ||
| Fig. 5 Representative arene–ruthenium complexes studied as active anticancer drug candidates. | ||
Studying the reaction of guanine derivatives, 9-ethylguanine (9-EtG) and others, with [(η6-arene)Ru(en)Cl]+ complexes is important for understanding the interaction of guanine and ruthenium metal.57,58,64,65 Studies show that the arene ligand and N–H hydrogen bonding (H-bonding) by the diamine (en) ligand may play a determining role in the stabilization of these metal–guanine adducts. The coordination of guanine to ruthenium in the complex [(η6-arene)Ru(en)(9-EtG)]+ (η6-arene = biphenyl (23), tha, dha) has been evidenced crystallographically.57 Guanine strongly binds through N7 to the metal centre where strong H-bonding between NH(en) and O–C(9-EtG) stabilizes the guanine adducts (Fig. 6).57,58 Replacement of the –NH hydrogen on en by a methyl group reduces the activity drastically. It is suggested that the presence of an –NH assisted hydrogen bond (H-bond) between the en and guanine is crucial for the activity of these complexes.64
 | ||
| Fig. 6 Chemical (left) and X-ray crystal structure of [(η6-biphenyl)Ru(en)(9-EtG)]2+ (23) showing the H-bond between NH(en) and O6(9-EtG), and the H-bond assisted network of the dimer (right). Adapted from ref. 57 with permission from the American Chemical Society. | ||
Replacing the flexible en ligand in [(η6-arene)Ru(en)X]+ (X = Cl, I), with rather bulky N,N donor ligands, such as 2,2′-bipyridine and related species, results in a drastic decrease in the activity due to the loss of H-bonding in the absence of –NH bonds in the ligand.64 The hydrogen bonding is therefore crucial to obtain high activity, as observed for en based analogous complexes (well supported by X-ray crystallographic studies). Interestingly, arene–ruthenium complexes [(η6-arene)Ru(bpy(OH)O)Cl]+ [η6-arene = benzene, indane, biphenyl, p-terphenyl, tha (20), dha, tetrahydronaphthalene (thn)] containing deprotonated 2,2′-bipyridine-3,3′-diol (bpy(OH)O) exhibit a significant enhancement in the anticancer activity compared to the analogous complexes containing bipyridine.36 The most active complexes contain arene with extended rings [(η6-arene)Ru(bpy(OH)O)Cl]+ (η6-arene = tha (20, Fig. 5) and tetrahydronaphthalene (thn)). In the crystal structure of [(η6-tha)Ru(bpy(OH)O)(9-EtG)]+ a strong CH⋯π interaction was also observed between the C–H of the extended arene and planar pyridyl rings of the bipyridine, whereas no π–π interactions were observed between the arene and 9-EtG, which is known to stabilize DNA adducts. Although, these complexes show strong coordination with 9-EtG, they do not show binding with DNA. In contrast to the 2,2′-bipyridine based complexes, arene–ruthenium complexes in which the ligand system incorporates a nitro group either at the m- or p- positions such as N-4-nitro-Ph-quinlinamide (21) and N-4-nitro-Ph-picolinamide (22) exhibit very promising cytotoxicity against a series of human tumor cell lines comparable to cisplatin and carboplatin.66 These complexes also interact with DNA and form adducts with guanine nucleotides.
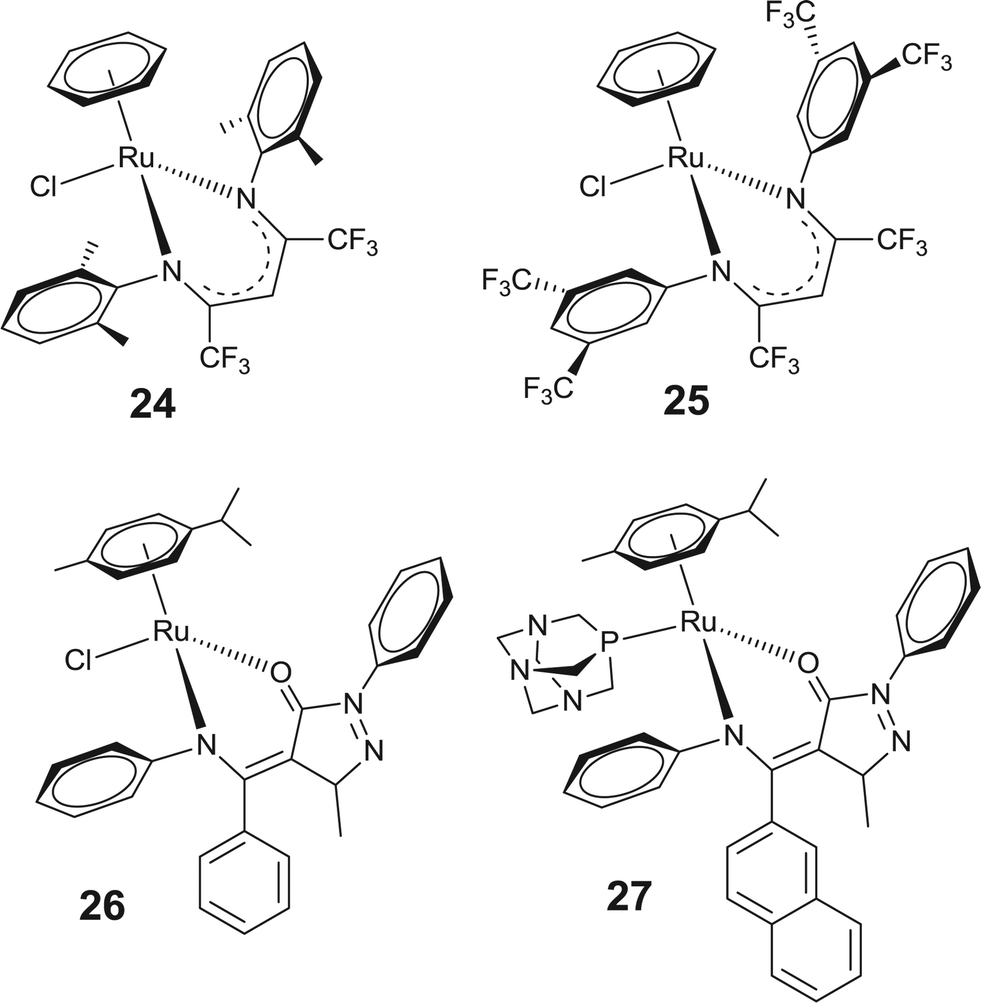
Recently, an interesting series of arene–ruthenium complexes based on β-diketiminato ligands has been evaluated for their in vitro anticancer activity.67 Among these the arene–ruthenium complexes [(η6-C6H6)Ru(β-diketiminato)Cl] with –CF3 substituents on the β-diketiminato = 2,6-(CH3)2C6H3NC(CF3)2CH, (24) ligand exhibited high cytotoxicity (Fig. 7). Furthermore, an enhancement in the cytotoxicity results in an increase in the number of –CF3 substituents on the β-diketiminate backbone and therefore the complex 25 shows cytotoxicity which is comparable or even superior to that of cisplatin. However, the authors of the article emphasized that the enhanced cytotoxicity does not emanate from the release of the cytotoxic β-diketiminato ligand because the loss of the β-diketiminato ligand was not observed under any conditions. Additionally, it has been observed that in these complexes the chloro group undergoes facile aquation without loss of the β-diketiminato ligand. Most recently, cytotoxicity of the β-diketoamine based arene–ruthenium complexes has also been investigated, where substituents of the β-diketoamine based acylpyrazolone ligand significantly controls the cytotoxicity of these complexes against human cancer (A2780) cell lines. Complexes with phenyl substituted β-diketoamine (26) exhibit higher cytotoxicity than those with naphthyl substituted β-diketoamine. However, replacing the chloride by pta or methanol in the arene–ruthenium complex (27) with naphthyl substituted β-diketoamine displays an enhancement in the cytotoxic activity.68
 | ||
| Fig. 7 Representative β-diketiminato based arene–ruthenium complexes showing high anticancer activity. | ||
Compared to N,N donor ligands, arene–ruthenium complexes containing N,O and O,O donor ligands exhibit only moderate activity with preferential coordination to the guanine base.69–71 Amino acid coordinated arene–ruthenium complexes [((η6-p-cymene)Ru(glycine)-N,O)Cl]+, presents biologically active complexes, but with lower cytotoxicity than analogous en complexes.64 The low cytotoxicity of these complexes presumably arises due to the high stability of the aqua complex of these amino acid adducts of the arene–ruthenium complexes relative to the respective chloro complex. Interestingly, [((η6-p-cymene)Ru(glycine)-N,O)Cl]+ rapidly reacts with 9-EtG in D2O to form the 9-EtG adduct of this complex [((η6-p-cymene)Ru(glycine)-N,O)(9-EtG)]2+ (28), by replacing the chloro group.64 Analogous to en complexes, the X-ray structure of this complex revealed the coordination of 9-EtG to Ru centre via N7 and strong H-bond formation between the ligand NH and O–C of the 9-EtG (Fig. 8).64 Despite the structural similarity between the arene–ruthenium complexes of en and N,O donor amino acids, the poor activity of the latter complexes is surprising. The observed low activity may be correlated with the fast hydrolysis rate, large reactive aqua species favor fast deactivation before reaching their target site.
 | ||
| Fig. 8 Chemical (left) and X-ray crystal structure of [(η6-p-cymene)Ru(glycine)(9-EtG)]2+ (28), showing H-bond between the NH(glycine) and O6(9-EtG) (right). Adapted from ref. 64 with permission from the American Chemical Society. | ||
Neutral arene–ruthenium complexes involving an acetylacetonato group (acac), e.g., [(η6-p-cymene)-Ru(H3CCOCHCOCH3)Cl] or its derivatives represent a class of potent arene–ruthenium complexes containing O,O donor ligands. In general, these display poor aqueous solubility and undergo hydrolysis to form an aqua adduct [(η6-p-cymene)-Ru(H3CCOCHCOCH3)(H2O)]+ more rapidly than those obtained with en ligands.34,64,70 The low potency of these complexes is due to easy protonation and the possibility of irreversible displacement of the chelated acac derivatives.34 In this respect, coordination of the sterically bulky acac ligand enhances the activity of these complexes to some extent by blocking the easy access of biomolecules to the Ru–metal center and therefore slows down the deactivation of the complex.70 However, these complexes display weaker binding with the nucleobase guanine relative to the analogous en complexes. The X-ray crystal structure of [(η6-p-cymene)Ru(Phacac)(9-EtG)]+ (29) (Phacac = PhCOCHCOPh) revealed a large C–O (9-EtG) to O (Phacac) bond distance (Fig. 9a), perhaps due to the repulsion between these oxygen atoms resulting in a decrease in the affinity of these acac containing arene–ruthenium complexes towards guanine, compared to the en analogues.70 However, unlike the arene–ruthenium complexes of en, the acac containing complexes also form stable systems with adenine [(η6-p-cymene)Ru(acac)(9-EtA)]+ (30) (Fig. 9b).70 The X-ray crystal structure shows a strong H-bonding between N6–H (adenine) and O (acac), which stabilizes the adenine adduct of arene–ruthenium complexes.
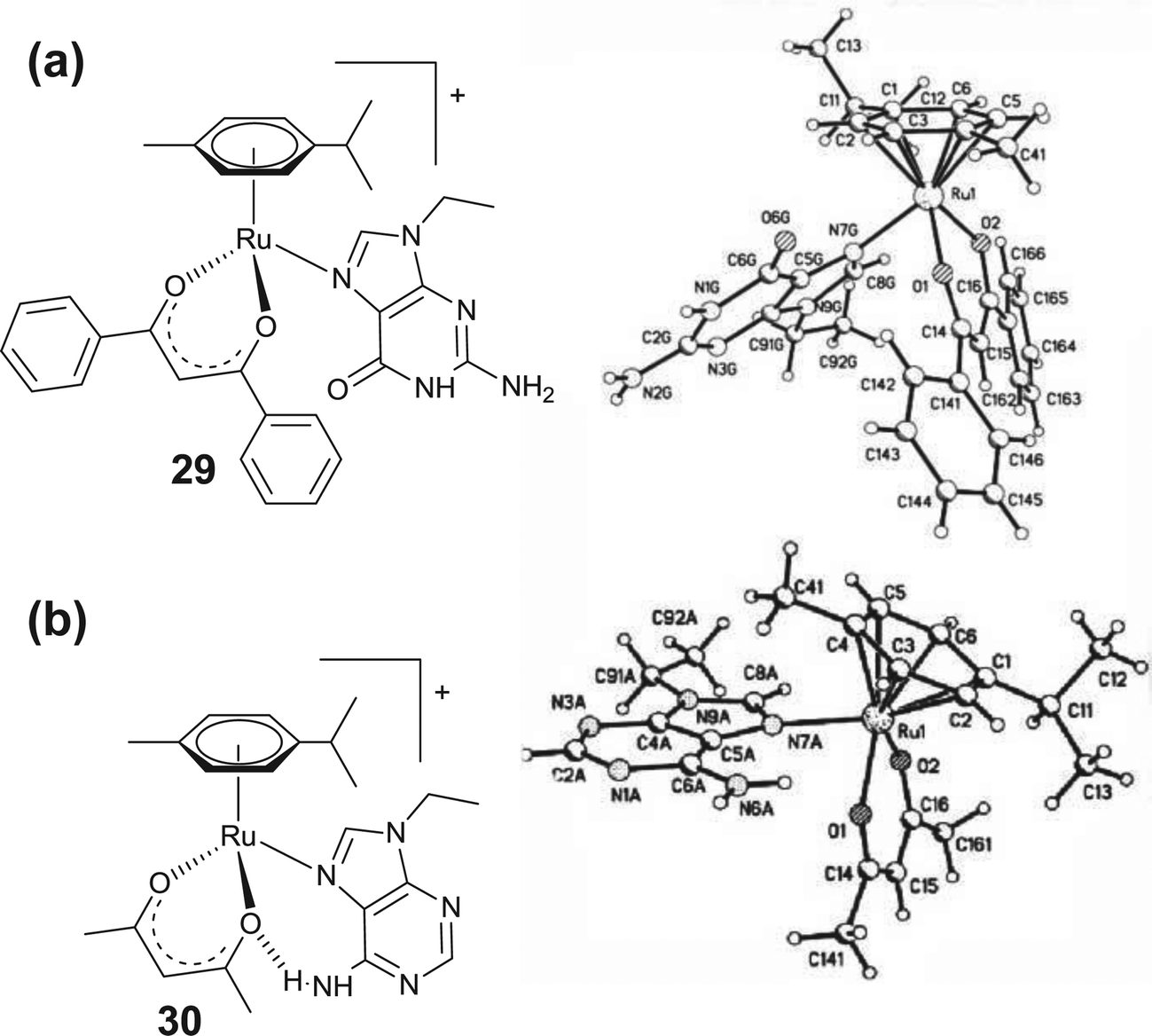 | ||
| Fig. 9 Chemical (left) and X-ray crystal structures of (a) [(η6-p-cymene)Ru(Phacac)(9-EtG)]+ (29) and (b) [(η6-p-cymene)Ru(acac)(9-EtA)]+ (30) complexes (right). Reprinted from ref. 70 with permission from Elsevier. | ||
Recently, Sadler and co-workers extensively reviewed arene–ruthenium complexes with special emphasis on the arene–DNA intercalation and the effect of these interactions on the mechanism of anticancer activity and structure–activity relationships.72 Studies revealed that various crucial processes like conformational distortions, recognition by DNA-binding proteins, and repair mechanisms are also dependent on the arene.73,74 Reports show that the cytotoxicity of the arene–ruthenium based drug candidates can be enhanced by using substituted arenes or arenes with extended π-systems which provides a hydrophobic face for the complex and enhances biomolecular recognition.69,75 For instance, the adducts of [(η6-p-cymene)Ru(en)]2+ can be removed from DNA more efficiently than those of [(η6-tha)Ru(en)]2+. The removal of the DNA–complex adducts follows a mechanism other than nucleotide excision repair, which is a major mechanism contributing to cisplatin resistance.33,76 Compared to non-intercalating p-cymene, the extended π-electron ring system of the biphenyl facilitates hydrophobic π–π stacking interactions of the arenes with DNA bases.77 In general, arene–ruthenium complexes with a benzene ring fused to a five, six or seven membered cyclic hydrocarbon, such as dha, tha, exhibit more favorable π–π stacking between arene and DNA bases.33,57,58,64 The activity increases with an increase in the size of the coordinated arene: benzene < p-cymene < biphenyl < dha < tha, and the leaving group which is typically chloride, can control the tuning of activation of these complexes.10,29 The enhanced potency may be related to the degree and ease of hydrophobic interaction of arene with nucleobases. The crystal structure of the arene–ruthenium complexes [(η6-arene)Ru(en)(9-EtG)]2+ with tha (31) and dha as the η6-arene shows a strong π–π intramolecular stacking between the pendant and the six-membered guanine ring with centroid-to-centroid separations of 3.45 Å for 31 (Fig. 10) and 3.31 Å for [(η6-dha)Ru(en)(9-EtG)]2+. The molecular arrangement of these complexes suggest simultaneous coordination of guanine to ruthenium and intercalation of the extended arene ring between the DNA base pairs, which are responsible for the observed high activity of these complexes.57
 | ||
| Fig. 10 Chemical and X-ray crystal structure of [(η6-tha)Ru(en)(9-EtG)]2+ (31) showing the intramolecular arene–guanine base stacking and H-bonding interactions between NH(en) and O6(9-EtG). Adapted from ref. 57 with permission from the American Chemical Society. | ||
Moreover, various arene–ruthenium complexes containing planar N,N chelating ligands also enhance the biological activity by intercalating between the base pairs of DNA. Lippard and co-workers demonstrated that metal complexes with planar aromatic coordinating ligands may bind DNA by intercalation where the planar part of the ligand undergoes non-covalent stacking interaction with DNA base pairs.78,79 These complexes interact with DNA preferably with its chromophore intercalated between the DNA base pairs. Thus, tuning the properties of the planar ligand can provide a versatile and an improved biological probe. Recently, arene–ruthenium complexes coordinated with phenanthroimidazole derivatives have been investigated. Among the studied complexes, those having -p-NMe2C6H4 substituents (32) display significant inhibition activities against several cancer cells (Fig. 11). The observed activity is mostly because of the cell cycle arrest at the S-phase which is induced by complex triggering of DNA damage mediated by p53 phosphorylation. Studies reveal that 32 interacts with DNA by an intercalative mode (through its extended planar portion) to disturb the bio-function of cancer cells.80
 | ||
| Fig. 11 Representative arene–ruthenium complexes showing interacalative mode of interaction with DNA. | ||
In addition, dual-function arene–ruthenium complexes introduce new mechanisms of antitumor activity, novel mechanisms for attack on DNA, and new concepts for developing structure–activity relationships (Fig. 12).72 The presence of extended arene moieties is an added advantage for the arene–ruthenium complexes which also show binding with DNA nucleobases (facilitated by aquation). The possible π–π stacking interactions between these arene and the DNA base pairs may result in a concomitant increase in the anticancer activities of these complexes. Such dual function arene–ruthenium complexes e.g., [(η6-arene)Ru(en)Cl]+ (η6-arene = biphenyl (3), tha (17)) are more potent towards cancer cells than their non-intercalating analogs such as [(η6-arene)Ru(en)Cl]+ (η6-arene = p-cymene (15), benzene)29 and less intercalating isomers (p-terphenyl (19) are more potent than o- or m-terphenyl complexes).35,81 In complex 19, the extended phenyl rings provide an opportunity for complex 19 to intercalate between the DNA base pairs as well as to undergo monofunctional binding to DNA preferentially, to guanine, in contrast to the monofunctional coordination of o- or m-terphenyl complexes to DNA bases. Low mutagenicity and different modes of action (intercalation and coordination) compared to Pt drugs (cisplatin), makes these drug candidates suitable for chemotherapeutic studies for human cancer. Furthermore, linking the intercalating moiety of naphthalimide to the arene–ruthenium complexes, 33 and 34 also leads to high cytotoxicity against human ovarian cancer cell lines.82,83 RAPTA-type complexes (33) containing a pendent naphthalimide unit which is tagged via an aliphatic chain to the arene ligand are slightly more active than those containing an imidazole linked naphthalimide unit (34) due to the presence of pta (Fig. 11). However, the additive property of the pta is presumably absent in complexes having imidazole linked naphthalimide. Studies reveal that naphthalimide mediated intercalation (π-stacking with guanosine) is preferred over direct coordination of the guanosine to ruthenium.82 However the ruthenium unit may bind to protein and therefore the observed dual mode of interaction (intercalation by naphthalimide and coordination of ruthenium with protein) is presumably responsible for the increased cytotoxicity shown by these complexes in comparison to RAPTA-C.
 | ||
| Fig. 12 Pictorial representation of dual mode of interaction (interacalation and binding) of [(η6-p-terphenyl)Ru(en)Cl]+ (19). | ||
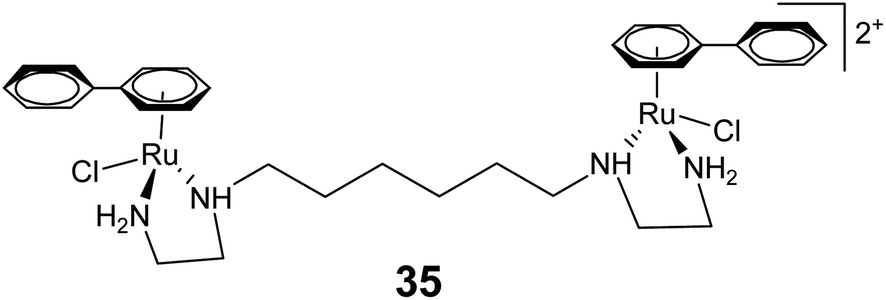
The dinuclear arene–ruthenium complex [{(η6-biphenyl)RuCl(en)}2-(CH2)6]2+ (35) (Fig. 13) can undergo double intercalation into DNA via induced-fit recognition involving epimerization at the dynamic stereogenic centers.84 It results in DNA cross-linking and generates interstrand cross-links. The intercalation properties displayed by 35 are significantly higher than their mononuclear analogs, because both of the free phenyl rings can contribute to the DNA intercalation for 35.
 | ||
| Fig. 13 Representative dinuclear arene–ruthenium complex showing double intercalation with DNA. | ||
3. Interaction of arene–ruthenium complexes with biomolecules (other than DNA), and arene–ruthenium complexes as biocatalysts
In the previous section we discussed the interaction of arene–ruthenium complexes with DNA nucleobases and correlated the biological activities and deactivation of the arene–ruthenium complexes to the rate and the mode of interaction of these complexes with DNA and its components. Recent research activities pointed out that for a clear understanding of the activities of these complexes, one has to explore their specific interactions and pathways with biomolecules other than DNA, within or outside the cell such as in extracellular matrices, biological fluids, cell surfaces.18–20 In this context, it has become crucial to solve protein mediated mechanisms, and to some extent, drug-resistance problems which include reduced cellular uptake, efflux of the drugs from the cells and so on.19 Unlike cisplatin, structural modification of the arene–ruthenium complexes can greatly suppress the drug-resistance mechanism. In this section we discuss the interaction of arene–ruthenium complexes with biomolecular targets other than DNA, e.g. protein, and biorelevant catalytic activities of the arene–ruthenium complexes.Recent findings on enzyme binding and targeting of biologically relevant proteins by arene–ruthenium complexes have stimulated new insight into understanding the activity of complexes for which DNA is not a target molecule.18–20,85 For example, RAPTA-C (4) exhibits a lower cytotoxicity than cisplatin, but high reactivity and selectivity towards protein binding relative to cisplatin. Messori and co-workers studied the inhibitory activity of a series of RAPTA complexes against thioredoxin reductase (TrxR) and cathepsin B (Cat B) as possible targets for anticancer metallodrug candidates.86 Almost all the studied RAPTA complexes show inhibition activity against Cat B, whereas poor to very low activity against TrxR. Moreover, the inhibition activities vary significantly for various RAPTA complexes. Docking experiments revealed that the inhibition activity of the RAPTA complexes (36) with Cat B was initiated by aquation of one chloride ligand and subsequent coordination of the ruthenium(II) center to the active site cysteine residue (Fig. 14). Furthermore, another chloride ligand along with a nitrogen atom of the pta or substituents on the arene ligand show close interaction with the amino acid residues of Cat B, thereby stabilizing the RAPTA–enzyme adduct (37). Analogous inhibition activity against TrxR and Cat B has been induced by arene–ruthenium complexes (38) containing N-heterocyclic carbene (NHC) (Fig. 14). These complexes preferentially exhibit high inhibition of TrxR, whereas inhibition of Cat B depends on the exposure time. The pronounced inhibition shown by these complexes is due to their interaction with biologically relevant thiols and selenols.87
 | ||
| Fig. 14 Representative arene–ruthenium complexes based active inhibitors. RAPTA complex showing main interaction with the active sites of cysteine ligand by replacing one chloride ligand and other close interactions with amino acid residues. | ||
Glutathione transferase (GST) which is found in solid tumors has also been identified as a possible target for antitumor drugs because of its over expression in the presence of antitumor drugs. GST is involved in the detoxification of antitumor drugs by catalysing the conjugation of glutathione (GSH) and antitumor drugs.88 RAPTA derivatives coupled with ethacrynic acid (EA), an effective GST inhibitor and a potential anticancer drug candidate, represent an active group of arene–ruthenium complexes (39 and 40) showing a dual cytotoxic mode of action (Fig. 15).89 These complexes have the combined effect of EA and RAPTA, and are therefore able to bind not only to the enzyme at the hydrophobic (H-site) co-substrate (speciality of EA) but also to interact with the reactive cysteine residues of GST (from RAPTA). For complexes where EA is linked with the amide bond of the arene (39), GST inhibition was found to be stronger. Studies revealed that these complexes coordinate with the cysteine residue of GST by loss of one chloride ligand. However, over a period of time, the complex decomposed into a ruthenium derivative and EA, and thus release of the ruthenium unit enhances the toxic effect. This report features an interesting example of the specific delivery of cytotoxic drugs for targeted chemotherapy.
 | ||
| Fig. 15 Representative arene–ruthenium complexes based enzyme inhibitors. | ||
Phenoxazine and anthracene derivatives exhibit excellent inhibition for P-glycoprotein (Pgp), which is a plasma membrane protein responsible for drug effluent from cells and is involved in multidrug resistance. The phenoxazine and anthracene based multidrug resistance modulator has been introduced by linking it with imidazole. The arene–ruthenium complexes, [(η6-p-cymene)Ru(N-imd-R)Cl2] (41) (N-imd-R = N-phenoxbenzimidazole and N-anthramidazole) with these structurally modified derivatives exhibit more cytotoxic action and even stronger Pgp inhibition. The enhanced activity is due to synergistic interaction of the arene–ruthenium unit with phenoxazine and anthracene derivatives, where the imidazole ligand acts as a linker to introduce multidrug resistant modulators by inhibiting DNA synthesis (Fig. 15).90
Kinetically inert half-sandwich ruthenium complexes have shown excellent and selective inhibition of protein kinases. Kinase, which catalyzes the important cellular function of phosphate group transfer for ATP to various substrates is a crucial target for antitumor drugs. In these complexes, coordination of the organic molecule to a metal centre provides stability and opportunity to get access to the unique chemical space which otherwise cannot be achieved by the organic compound itself.19,20 The inert cyclopentadienyl–ruthenium complex (42 and 43) with a pyridocarbazole ligand, is a derivative of a non-selective kinase inhibitor and the natural product staurosporin, and represents an active class of protein kinase inhibitors whereas the pyridocarbazole ligand exhibits only limited selectivity (Fig. 15).91,92 These complexes inhibit protein kinases by competitively binding to the ATP binding sites of the enzyme. Co-crystallization of 43 with human protein kinase Pim-1 (Proto-oncogene serine/threonine–protein kinase)93 and PAK1 (p21-activated kinase 1)94 illustrates that the half sandwich ruthenium complex closely mimics the binding mode of staurosporin, because the essential features of the planar indolo[2,3-a]carbazole aglycon of the staurosporin has been retained by the complex 43 (Fig. 15). In the 43–Pim1 adduct (Fig. 16), the imide hydrogen is involved in hydrogen bonding with the adenine binding cleft which plays a crucial role in the high activity of these complexes. Methylation of the imide position leads to deactivation of the complexes.93 Various hydrogen bonds and van der Waal contacts observed between the complex and ATP binding sites of Pim-1, resemble with the binding pattern of ATP well. In addition, the cyclopentadienyl ring and CO also show van der Waal and dipolar interactions with the aminoacid adducts of Pim-1, and therefore contribute to the observed high potency of these complexes as excellent kinase inhibitors. In a subsequent study, the involvement was demonstrated of the planar imide moiety and indole OH-group of the cocrystal of 43 with PAK1to mediate the hydrogen bonds to the backbone of the hinge region, and the carbonyl ligand was shown to interact with the amino acid residues.
 | ||
| Fig. 16 Chemical structure of staurospin (left) and interactions of 43 with ATP binding sites of Pim-1 (indicated by arrow) (right). | ||
Furthermore, derivatization of the ligand and the absolute configuration of these complexes has a significant effect on their selectivity towards targeting protein kinases.91 The S-isomer of the complex (43) containing an hydroxy substituent on the indole ring exhibits a 30-fold higher selectivity for Pim-1, whereas the R-isomer (42) having hydroxyl and bromo substituents on the indole and methoxycarbonyl groups on the cyclopentadienyl ring is more selective for GSK-3 (glycogen synthase kinase 3) (Fig. 15). These also effectively target lipid kinases, found in a variety of human cancers.95 Despite the structural homology of the lipid kinase, such as PI3K (phosphatidyl-inositol-3-kinase), and protein kinases, the binding is quite divergent to that with protein kinases. For instance, direct hydrogen bonding interactions by imide hydrogen are not present in PI3K, which is inferred by an enhancement of inhibition activity by methylation of the maleimide imide. However, unlike the metallo-pyridocarbazoles, 42 and 43, the position of the metal center relative to the ATP binding sites has a very drastic effect on the binding profile and therefore, these complexes are probably suitable for only a limited number of protein kinases, whereas metal complexes with pyridylnaphthalimide exhibit very promising results.96 The neutral complex 44 inhibited proliferation of the melanoma cell lines in a dose-dependent fashion (Fig. 15). The observed activity has been attributed to the interaction of 44 with the hinge region of ATP binding sites by forming two hydrogen bonds with imide moiety.
Efficient catalytic oxidation of the major intracelluar reducing agent GSH to its disulfide form (GSSH) is of significant importance in the activity of anticancer drugs.97 Since, cancer cells have a higher GSH pool than healthy cells, an increased GSSH-to-GSH ratio is indicative of oxidative stress which can damage all cellular components and lead to apoptosis. The half-sandwich arene–ruthenium complex 45 (Fig. 17) containing N,N donor phenylazopyridine (azpy) ligand [(η6-arene)Ru(azpy)X]n+ (X = Cl, I) exhibits high cytotoxicity against cancer cells following a new mechanism of drug action (for instance, chlorido complexes are taken up by means of active transport, whereas iodido enters cells via passive transport).98–101 Remarkably, certain choices of the azpy derivative and monodentate ligand (X), particularly the iodo (I−) ligand, give rise to complexes which are inert to aquation and yet exhibit high activity towards both ovarian and lung cancer cells. It has been presumed that these complexes get activated in cells by reduction of the azpy ligand with subsequent generation of the reactive oxygen species which oxidise the tripeptide glutathione to its GSSH. Suss-Fink and co-workers also reported a highly cytotoxic water soluble diruthenium complex [(η6-p-cymene)2Ru2(SC6H4-p-R)3]+, R = Me (46) and But, (47) (Fig. 17), which catalyzes the oxidation of thiols cysteine and GSH.102 The tested diruthenium complex performed well with the oxidation of cystein thiols into free cysteine and GSH to GSSH. Since the intracellular GSH content in human cancer cell (A2780) lines are higher, the observed excellent catalytic oxidation of GSH is of significant importance and is reflected in the high cytotoxicity of this complex against A2780 cancer cell lines (complex 47 exhibits very high cytotoxicity). The observed high cytotoxicity of the studied complexes is presumably due to the physicochemical properties of the complexes as determined by the lipophilicity of the thiols and electronic behavior of the substituents.
 | ||
| Fig. 17 Representative catalytically active arene–ruthenium anticancer complexes. | ||
Sadler and co-workers investigated the possibility of using arene–ruthenium complexes for catalytic NAD+/NADH hydride transfer reactions in cells as a possible novel mechanism of action. Both NAD+ and NADH play important role as cofactors in many bio-catalytic reactions, energy metabolism, immunological functions, and apoptosis.103–105 Using formate as the hydride donor, catalytic regioselective reduction of NAD+ can be executed in the presence of complex 48 (Fig. 17). However, the feasibility of the catalytic reaction is due to tolerance of the target cells for formate.106 Later studies revealed that by replacing en with bpy or phen, the reverse catalytic hydride transfer from NADH to NAD+via the metal centre can be performed.107 Moreover, complexes having strong electron donating substituents such as hexamethylbenzene in 49, facilitates the catalytic NADH to NAD+ conversion (Fig. 17). These results inferred that the studied complexes can mimic enzymes such as lactate dehydrogenase which use NADH as a cofactor.
4. Photoactivated arene–ruthenium drug candidates
One of the crucial steps in the activation of ruthenium complexes as potential anticancer agents is usually aquation of the Ru–X bond (X = labile monodentate ligand) to form a reactive aqua species.44,108 In this context, photochemical activation is an attractive tool to control the biological activity of arene–ruthenium complexes, which are in general inactive in the dark. Sadler and co-workers explored photo controlled activation of the arene–ruthenium complex [(η6-p-cymene)Ru(bpm)(py)][PF6]2+ (50, bpm = 2,2′-bipyrimidine and py = pyridine). An aqueous solution of this complex is stable in the dark while the Ru–pyridine bond is dissociated in the presence of visible light thereby releasing the monodentate pyridine ligand. Upon photo-irradiation, both the Ru → bpm and Ru → py states get excited, but the strong coordination of bipyrimidine by other pyrazines prevented Ru–bpm bond dissociation, rather than the relatively facile Ru–pyridine bond dissociation.109 Therefore, this mode of activation facilitates the coordination of newly generated reactive aqua species (51) to the DNA base guanine (Fig. 18).109 Interestingly, covalent linkage of a delivery peptide to this arene–ruthenium complex through pyridine improves the selectivity of these complexes against cancer cells.110 Upon photolysis with visible light, the ligand dissociation process was initiated and released both the entities, the peptide linked pyridine and activated aqua species. The presence of the peptide fragment facilitates the cellular uptake of the activated arene–ruthenium complex by improving its aqueous solubility, where the active aqua species reacts with DNA. This strategy of generating photoactivated arene–ruthenium based drug candidates provides a dual mode of action where the stable pro-drug can be photochemically activated showing controlled activity and targeted selectivity. An analogous trend of photoactivation has also been reported by Sadler and co-workers in their detailed investigations on [(η6-p-cymene)Ru(bpm)(L)]2+ by using a variety of monodentate ligands (L), where selective dissociation of the monodentate ligand facilitates their preferential binding with 9-EtG and 9-EtA to form [(η6-p-cymene)Ru(bpm)(9-EtG)]2+ (52).111 | ||
| Fig. 18 Arene–ruthenium complex showing selective photoactivated dissociation of monodentate ligand (pyridine) and coordination to the guanine base of DNA. | ||
Most recently, the arene–ruthenium complex (53) containing dipyrido-[3,2-a:2′3′-c]phenazine (dppz) as the planar chelating ligand has been reported to display high photocleavage activity towards DNA.112 A 10 fold enhancement in the activity was observed for 53 upon visible light activation compared to the activity in darkness. However, replacing the ligand dppz with bpy leads to an inactive complex, thereby supporting the significance of the dppz ligand as a photosensitive unit. Furthermore, photoactivation studies on dinuclear arene–ruthenium complexes [{(η6-indane)-Ru(Cl)}2(μ-2,3-dpp)2]2+ (54, dpp = 2,3-bis(2-pyridyl)pyrazine) revealed a different mode of activation (Fig. 19).113 The dinuclear complex 54 may undergo aquation in dark, but exhibits poor binding activity towards DNA. Irradiation of the DNA–complex adduct or irradiation of the complex in the presence of DNA led to an increased interaction between DNA and the complex. It has been observed that the dinuclear complex generates a new active ruthenium species by arene loss upon irradiation. Moreover, generation of active ruthenium species by photoactivation of the dinuclear arene–ruthenium complex also depends on the nature of the arene moiety, where the benzene analogue also undergoes photoinduced arene loss, but the strongly coordinated arene such as hexamethylbenzene remains intact.
 | ||
| Fig. 19 Representative arene–ruthenium complexes showing photo-induced anticancer activity. | ||
The combination of a photosensitizer and a chemotherapeutic agent such as the arene–ruthenium complex represents a promising class of light driven biologically active agents. Activation of the photosensitizer by light such as in ruthenium–porphyrin leads to production of reactive oxygen species responsible for photocytotoxicity.114,115 Coordinating arene–ruthenium units to a porphyrin results in effective light triggered therapeutic agents against human cancer cells (Fig. 20). Porphyrin coordinated arene–ruthenium complexes [{(η6-p-cymene)RuCl2}4(TPP)] (TPP = 5,10,15,20-tetra(4-pyridyl)-porphyrin and 5,10,15,20-tetra(3-pyridyl)porphyrin) exhibit only moderate to no cytotoxicity in the dark against melanoma cancer cells. However, upon light exposure of these complexes, a 60–80% increase in the phototoxicities has been observed.114 Presumably, an increase in the nuclearity favours an increase in the solubility of the highly hydrophobic porphyrin ligands in polar organic solvents and thereby may enhance the uptake of porphyrin by human melanoma cells.9,114 Moreover, the nature of the pyridylporphyrin isomers has a distinct effect on the photodynamic properties of the complex and therefore the 3-pyridyl-porphyrin containing complex (55) had a better photodriven activity relative to the 4-pyridylporphrin analogues.116 Interestingly, the tetranuclear complex 55 has a higher activity than its mononuclear analogue due to the additive effect that arises from the coordination of photosensitizers (porphyrin) to arene–ruthenium.116 The observed light driven enhancement of the cytotoxicity of porphyrin containing arene–ruthenium(II) complexes may be due to the synergistic effect of the photosensitising properties of porphyrin and the chemotherapeutic effect of arene–ruthenium complexes.116
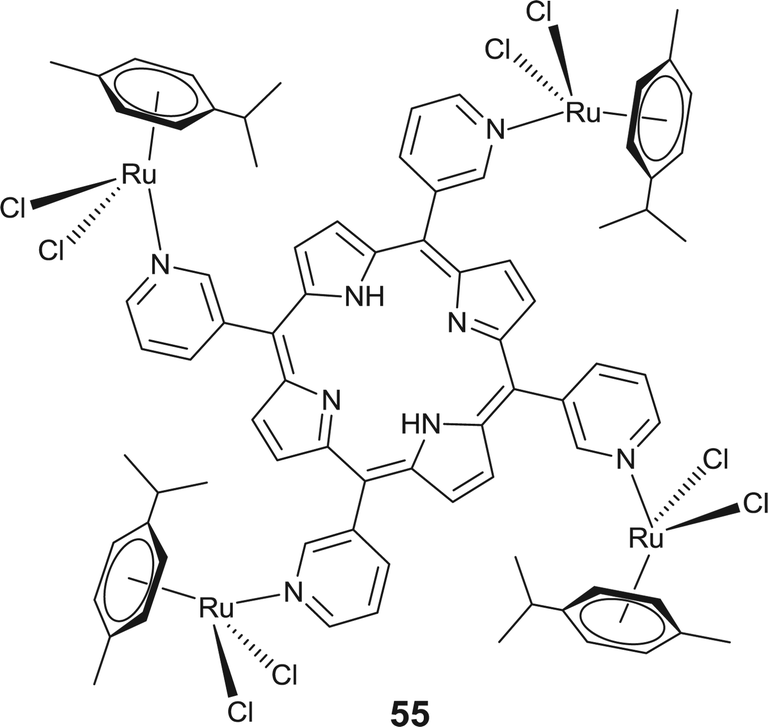 | ||
| Fig. 20 Representative cytotoxic arene–ruthenium complex containing porphyrin based photosensitizing agent. | ||
In an alternative approach, photosensitizers such as porphyrin can be delivered to cancer cells using water-soluble metallacages for targeted cytotoxicity.117 Water soluble arene–ruthenium metallacages have been used to deliver cytotoxic molecules to cancer cells (discussed in later sections). The encapsulation of the lipophilic photosensitizer, porphyrin, in two cationic arene–ruthenium metallacages [(η6-p-cymene)6Ru6(tpt)2(dobq)3]6+ (56, tpt = 2,4,6-tri(4-pyridyl)1,3,5-triazine; dobq = 1,4-benzoquinone-2,5-diolato) and [(η6-p-cymene)8Ru8(tpvb)2(donq)4]8+ (57, tpvb = 1,2,4,5-tetrakis{2-(4-pyridyl)vinyl}benzene; donq = 1,4-naphtoquinone-5,8-diolato) facilitates the intracellular delivery of the photosensitizers (Fig. 21). In contrast to the trapping of porphyrin in the cavity of 56, the metallacage 57 exhibits a reversible encapsulation of the porphyrin. Fluorescence experiments show that the release of porphyrin is higher for the larger cubic cage 57 as compared to the small prismatic cage 56. Excellent photocytotoxicity observed for these metallacages confirms the release of porphyrin from the cage. Furthermore, the 10 times higher phototoxicity of 57 relative to 56 is well supported by fluorescence results, suggesting the dependence of cytotoxicity with release of the porphyrin from the cavity. Most importantly, the encapsulated photosensitizer remains non-reactive to light during its transport to the intracellular matrix, suggesting that the safe and efficient delivery of photosensitizers by arene–ruthenium metallacages could be a new strategy for new photodynamic treatment.
 | ||
| Fig. 21 Representative metallacages encapsulating the photosensitizer, porphyrin, in the cavity. Adapted from ref. 117. | ||
5. Metal–drug interactions in arene–ruthenium complexes
An alternative approach to the discovery of new metallodrugs involves binding an organic compound of known therapeutic value to a metal-containing fragment. This concept of metal–drug synergy comprises a metal, which acts as a stabilizer for the drug until it reaches its target, while at the same time the organic drug protects the metal, preventing side reactions in its transit towards a target of biological action. The linking of biologically important organic molecules to arene–ruthenium species led to significant enhancement of cytotoxicity, by modulating the biological activity of the organic compound (Fig. 21).118–121Chloroquine, an effective organic antimalarial drug as well as having anticancer properties, coordinated arene–ruthenium complexes, such as [(η6-p-cymene)RuCl2(η1-chloroquine)] (58), represent a potentially active group of complexes which show higher activity than chloroquine (Fig. 22).122 In complex 58, chloroquine binds to ruthenium in the η1-mode through the quinoline nitrogen, and displays high activity against P. falciparum. More significantly, the potency of all the complexes against chloroquine-resistant parasites is consistently higher than that of standard drug chloroquine diphosphate, demonstrating that the Ru–chloroquine combination is able to affect the parasite's resistance mechanisms. Analogous reports with arene–ruthenium coordination (59) with a pharmacophore, such as 3-(benzazol-2-yl)-1H-quinoxalin-2-ones, reveal an enhancement in the antiproliferation potency.123 The observed high activity of organic drug candidates upon coordination with a metal core is due to enhancement of their solubility, which maximizes the availability of the drug at the cellular level. Moreover, the metal–drug synergistic activity is also influenced by stability, inertness/reactivity and solubility of these complexes (Fig. 22).
 | ||
| Fig. 22 Representative cytotoxic arene–ruthenium complexes coordinated by biologically active organic drug candidates. | ||
Extending the concept of metal–drug synergy, a recent report shows that the combination of clotrimazole, a well known antifungal agent, with arene–ruthenium species (60) enhances the antiparasitic activity by 50–100 times relative to the free organic drug.124 The synergistic effect that arises from the combination of clotrimazole and the metal containing moiety in a single molecule significantly contributes to the observed high activity, and therefore the presence of a metal centre is critical for controlling the physicochemical properties of an organic drug. A recent report shows that complex 61 resulting from the coordination of lonidamine-modified imidazole ligand to an arene–ruthenium moiety, exhibits high cytotoxicity in human glioblastoma cell lines. The displayed high inhibition activity is due to the coordination of lonidamine, [1-(2,4-dichlorobenzyl)-1H-indazole-3-carboxylic acid], which inhibits aerobic glycolysis in cancer cells thus providing a potentially highly selective ruthenium drug molecule.125 Interestingly, 61 exhibits more cytotoxicity than lonidamine and RAPTA-C indicating the cooperative interaction between the two units. Furthermore, incorporation of the propyl-imidazole group into the lonidamine also enhances the overall cytotoxic effect which is further enhanced by coordination to the arene–ruthenium moiety. The complex 43 coordinated with a derivative of an active organic compound, staurosporine also acts as a potential protein kinase inhibitor, this is due to the retention of essential features of indolo[2,3-a]carbazole aglycon, staurosporine (Fig. 16).
Similarly, antibacterial agents such as quinolone and its derivatives, which are widely used in clinical practice (Fig. 23) are supposed to bind to DNA–topoisomerase complex thus preventing the bacteria from replicating.126,127 Turel and co-workers have extensively investigated arene–ruthenium complexes, [(η6-p-cymene)RuCl(nalidixicato-κ2-O,O)] (62) (Fig. 23) containing mportant biologically active organic molecules, such as ofloxacin, nalidixic acid and cinoxacin.128,129 Furthermore, they examined these complexes considering their drug-like properties such as stability in aqueous solution and reaction with biomolecular target molecules, as well as studying their tumor-inhibiting potential in a cancer cell line panel. It has been established that replacing the chloro ligand with a labile water molecule in these complexes enables the compounds to readily interact with target molecules such as DNA. Substituting the pyridine oxygen in these complexes by sulphur led to a [(η6-p-cymene)RuCl-(thionalidixicato-2-S,O)] complex which shows high stability in aqueous solution and improved cytotoxicity.130 Analogously, a new class of arene–ruthenium complexes with 2-substituted indoloquinolines (63, Fig. 23) displays remarkably high antiproliferative activities in human cancer cell lines. Binding to arene–ruthenium moieties resulted in improved solubility, whereas metal-free modified indoloquinolines display poor solubility, enabling most of them to be tested as potential antitumor agents.131 The high antiproliferative activity is presumably due to their potential to act as DNA intercalators.132 Furthermore, changing the binding moiety in 63, so as to keep the lactum moiety free does not lead to any significant improvement in the activity.133
 | ||
| Fig. 23 Biologically active organic molecules and their representative arene–ruthenium complexes. | ||
Naturally derived compounds such as flavonoid and others are also considered as promising candidates because they exhibit promising biological properties upon coordination to a metal.134 Recently the topoisomerase IIα inhibitory activity of the arene–ruthenium complexes containing flavonoid ligand [(η6-p-cymene)Ru(flavonoid)Cl] (64) was reported. These exhibit higher affinity towards the N7 atom of the guanine base, keeping intact the flavonol ligand.135 The activity of 64 appears to be influenced by substituents on the phenyl ring of the flavonoid based ligand where para- and meta-substituted ligands exhibit lower activity than their unsubstituted analogues or the ortho-substituted derivatives (Fig. 24).135,136 However, substituents on the arene only have a minor effect on the cytotoxicity.137 Interestingly, the antiproliferative activity of these complexes correlated well with the enzyme inhibition activity. Higher activity of the arene–ruthenium–flavonoid complex relative to that of the free organic ligand demonstrated the significance of the metal–drug synergy. Similarly, coordination of the bioactive lapachol [(2-hydroxy-3(3-methylbut-2-2n-1-yl)naphthalene-1,4-diene) with the arene–ruthenium moiety, results in a metal–organic adduct (65) with enhanced biological properties (Fig. 24). Upon facile hydrolysis, the activated complex interacts with biomolecules such as 9-EtG, amino acids (glycine, L-cysteine) and so on. However, coordination with the amino acids involves a two-step binding process, firstly the monodentate coordination of amino acid and then cleavage of the lapachol by labilisation of O,O-chelate. These complexes exhibit lapachol based cytotoxicity, which may be related to the ligand release in the presence of biomolecules.138 To explore the metal–drug synergism arene–ruthenium complexes based on the β–diketonate ligand such as curcumin, have also been reported (Fig. 24).139 Curcumin exhibits biological properties such as antioxidant, anti-inflammatory, anti-microbial and anticancer activity.140,141 Upon coordination of curcumin with arene–ruthenium species in [(η6-p-cymene)Ru(curcuminato)Cl] (66), a high in vitro activity for tumor cell lines has been observed due to metal–drug interaction. Complex 66 shows good antitumor activity against breast MCF7 and ovarian A2780 cell lines, comparable to other active groups of the arene–ruthenium complexes.
 | ||
| Fig. 24 Representative arene–ruthenium complexes with biologically active organic molecules. | ||
6. High-nuclear arene–ruthenium complexes as drug candidates and carriers
The “multinuclearity approach” has been explored extensively for arene–ruthenium complexes which has proved to be an important concept for ruthenium-based drug candidates.15,142–144 The high nuclear compounds have been evaluated for in vitro anticancer activity and it has been observed that a number of ruthenium centers and types of spacer used significantly influence the cytotoxicity of these metalla-assemblies. In this section we discuss the effect of an increase in nuclearity on the cytotoxicity of arene–ruthenium complexes. We also discuss the application of high-nuclear scaffolds as drug carriers and delivery systems.A significant enhancement may be expected with the increase in nuclearity, for example the dinuclear arene–ruthenium complex 35 exhibits significantly higher interaction with DNA than its mononuclear analogs.84 Recent findings on highly active dinuclear arene–ruthenium complexes have indeed confirmed this trend. The dinuclear arene–ruthenium complexes where two arene–ruthenium units are linked by a pyridine-derived linker show high cytotoxicity against human ovarian (A780) cancer cells. A systematic correlation between the linker size (lipophilicity) and cytotoxicity has been observed in these complexes, e.g., the complex 67 with a long chain (n = 32) is highly active (Fig. 25). The observed high activity is due to their ability to cross-link with biomolecules.145,146 Similarly, dinuclear arene–ruthenium complexes (68) with a ferrocenoyl pyridine based linker is almost twice as active as the mono-ruthenium analogue (Fig. 25).147 Sadler and co-workers also reported a dinuclear complex 54 (Fig. 19) which shows photodriven cytoactivity accomplished by the generation of a new active ruthenium species by arene loss upon irradiation.113 Suss-Fink and co-workers reported trithiophenolato-bridged dinuclear arene–ruthenium complexes [(η6-arene)2-Ru2(SC6H4-p-X)3]+, X = Me (46) and But (47) showing high activity against human ovarian cancer cell lines and is marked as one of the most cytotoxic arene–ruthenium complexes (Fig. 17).102 Over a broad range of cytotoxicities shown by these complexes, the thiophenyl and arene substituents exhibit a distinct effect on the cytotoxicity. The arene–ruthenium thiophenato complexes (69, Fig. 25) display systematically more cytotoxicity than their hydroxyl thiophenato analogues whereas no precise correlation has been observed between the effect of arenes and their lipophilicity.148 Recently, a series of trinuclear p-cymene ruthenium metallacycles connected with aminomethyl-substituted 3-hydroxy-2-pyridone ligands have been evaluated in vitro against cancer and fibroblast cell lines.149 It has been observed that water-soluble trinuclear complexes undergo fragmentation after uptake, thus giving rise to cytotoxic mononuclear complexes. Similarly, the porphyrin coordinated tetranuclear arene–ruthenium complex 55 exhibits high phototoxicity compared to its mononuclear analogue.114 The observed significant disparity in the activity is due to the beneficial effect arising from the coordination of arene–ruthenium moieties with porphyrin.
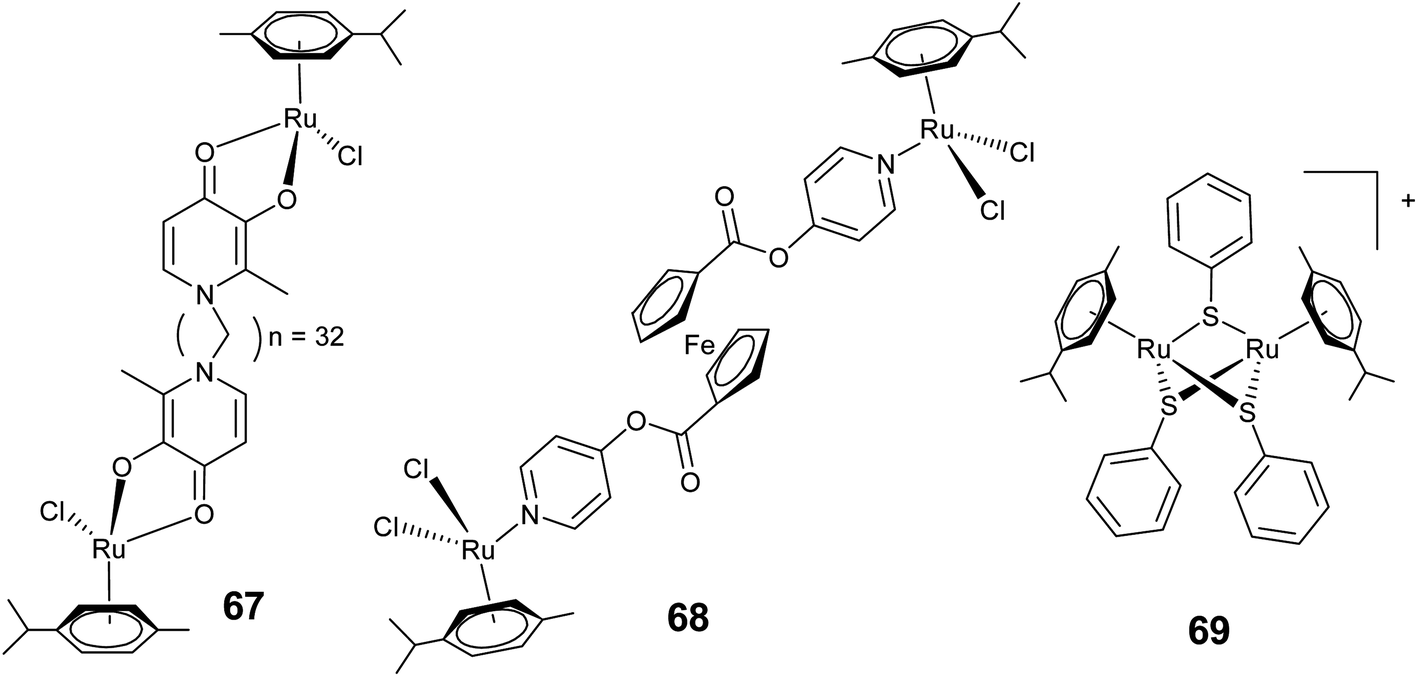 | ||
| Fig. 25 Representative dinuclear arene–ruthenium complexes showing high cytotoxicity. | ||
Therrien and co-workers extensively studied the antiproliferative activity of a series of water soluble arene–ruthenium tetranuclear metallarectangles (Fig. 26) [(η6-arene)4Ru4(L)2(N–N)2]4+ (η6-arene = p-cymene, hexamethylbenzene; L (oxygen bridged) = dobq, donq, and others; N–N (nitrogen bridged) = pyrazine, 4,4′-bipyridine, 1,2-bis(4-pyridyl)ethylene, 1,2-bis(4-pyridyl)ethane) against an ovarian cancer cell line.150,151 Interestingly, large metalla-rectangles 70 and 71 based on benzo- or naphthaquinone (R1) bridged arene–ruthenium containing 1,2-bis(4-pyridyl)ethylene (L1) and 1,2-bis(4-pyridyl)ethane (L2) as linkers exhibit higher cytotoxicity than those containing smaller linkers, such as 4,4′-bipyridine. Moreover, metallarectangle 72 with 4,7-phenanthroline (L3) linker displayed superior activity against human cancer cells due to the inherent intercalation shown by phenanthroline rings with DNA.152 It has been observed that the arene–ruthenium based metallarectangles can be taken up more efficiently by tumor cells, which are permeable to large, non-natural molecules, compared to healthy cells. This provides a degree of selectivity and ultimately led to reduced drug side effects.
 | ||
| Fig. 26 Representative arene–ruthenium complexes based metallarectangles and metallacages showing high cytotoxicity. | ||
A similar report by Chi and co-workers on arene–ruthenium based molecular rectangles containing an asymmetric donor, N-(4-(pyridin-4-ylethynyl)phenyl)-isonicotin-amide (L4), highlights the anticancer activity of multinuclear complexes (Fig. 26).153 Moreover, the naphthaquinolato based metallarectangles (73) exhibit higher activity than the oxalato based systems. Their recent studies on metallarectangles 74 and 75 showed that naphthaquinolato bridged dinuclear arene–ruthenium moieties linked with 1,2-di(pyridin-3-yl)ethyne (L5) and 1,4-di(pyridin-3-yl)buta-1,3-diyne (L6) are highly cytotoxic against human cancer cells compared to oxalate and benzoquinolato bridged analogues.154 Analogously, metallarectangles (76 and 77) based on azopyridyl linkers, (1,2-dipyridyl-4-yl)diazene (L7) and 1,2-bis(pyridine-4-ylmethylene)hydrazine (L8), exhibit high antiproliferative activity against cancer cells.155 Moreover, metallarectangles (78) based on naphthaquinolato bridged dinuclear arene–ruthenium moieties with dipyridyl amide linkers, N,N-bis(pyridine-4-yl)oxalamide (L9) exhibit high cytotoxicity comparable to cisplatin against human cancer cells, whereas the oxalate bridged analogous are inactive.156 However, analogous metallarectangles (79) based on bis-benzimidazole bridged arene–ruthenium moieties R2 display only moderate activity.157 Systematic in vitro investigations revealed that molecular rectangles with a high degree of aromaticity show a higher activity towards anticancer cells. The higher inhibitory effect shown by the metallarectangles on the growth of cancerous cells may arise from the induction of apoptosis. However, no direct correlation between metallarectangles size and cytotoxicity has been observed for these systems.
Stang and co-workers reported proliferative activity of arene–ruthenium metallaprismatic cages that were tested in vitro against several cancer cell lines (Fig. 26).158 Examining a series of metallaprismatic cages (80) based on donq bridged dinuclear arene–ruthenium fragments linked with ligands having varying π-conjugated systems, 1,3,5-tris(4-pyridylethynyl)benzene (L10) they found that the cytotoxicity of these systems are considerably higher than that of cisplatin.158 Recent reports on metallaprismatic cage 81 having bis-benzimidazole bridged arene–ruthenium moieties, demonstrated high cytotoxicity for this complex, compared to the inactive oxalate bridged analogue.157 The enhanced activity is due to the increase in nuclearity and the presence of an extended π-conjugated system. However, the observed activity was much lower than the naphthaquinolato bridged analogue.158 Their finding suggests that the reported arene–ruthenium based metallaprismatic cage is able to interfere with many parts of the regulatory pathways via apoptosis, which controls the cell growth cycle and shows that they could be useful for treating tumors which are generally difficult to recover by chemotherapy.
Moreover, efficient drug delivery to the cellular level is essential to fully exploit the therapeutic effect of a drug. Metallacages provide a highly efficient system for drug delivery, by avoiding pre-mature activation of the drug candidates and specific delivery to the target cells. Water-soluble metallaprisms synthesized from dinuclear arene–ruthenium complexes and tpt (L11) that can encapsulate planar aromatic molecules (e.g., pyrene, coronene),159 or square-planar complexes (e.g., [Pd(acac)2], [Pt(acac)2]),160 are a few interesting examples of drug delivery systems based on arene–ruthenium complexes. Exploiting the host–guest approach, Kim, Therrien and co-workers reported some arene–ruthenium based water-soluble organometallic cages to deliver floxuridine derivative drug to cancerous cells.161 Exploring a wide range of functionalized nucleic acid guests (pyrenyl nucleosides), the author of the article investigated antiproliferation effects of the host–guest combination model on ovarian cancer cells and its cisplatin resistant strains. Though the studied organometallic cages, [(η6-p-cymene)6Ru6-(tpt)2(dobq)3]6+ (56) and [(η6-p-cymene)6Ru6(tpt)2(donq)3]6+ (82) themselves show some activity, the overall activity drastically increased with the encapsulation of drug guest pyrenyl derivatives in the cavity. The high activity of water soluble organometallic cages provides stability to the host–guest system, while retaining the physical properties of the host cage. Furthermore, encapsulating the modified RAPTA complexes, with the inherent synergistic effect due to linking a DNA intercalator, such as pyrene to the arene [(η6-arene-pyrenyl)Ru(pta)Cl2] (pyrenyl–RAPTA),162,163 in the hydrophobic cavity of the large water-soluble metallacage 82 ensures effective cellular uptake of these arene–ruthenium complexes and achieves effective and targeted cytotoxicity (Fig. 27).164 These water soluble high-nuclear arene–ruthenium assemblies effectively deliver these pyrenyl linked RAPTA series of complexes to the cellular level. The metallacages 56 and 57 were also found to be suitable for encapsulating photosensitizers such as porphyrin to deliver photosensitizers to the target cells and exhibit photodriven specific cytotoxicity.117 The size of the cavity in these metallacages determines release of the photosensitizers. Metallacage 56 trapped the porphyrin in its smaller cavity whereas 57 having a large cavity shows reversible encapsulation of the porphyrin. Moreover, larger metallacages (83) have been prepared using tetra(pyridyl)porphyrin instead of tpt by linking with dobq or oxalato bridged dinuclear arene–ruthenium fragments (Fig. 27). Linking porphyrin rings by coordination bonds in the studied metallacages (83), hindered the intercalative mode of interaction of porphyrin but its binding ability was retained. These metallacages exhibit strong interaction with quadruplex DNA, with moderate selectivity.143 These interactions are reflected in the high cytotoxicity shown by these metallacages against A2780 and A2780cisR cancer cells. Introduction of a metal in the porphyrin ring afforded even more cytotoxic metallacages.165
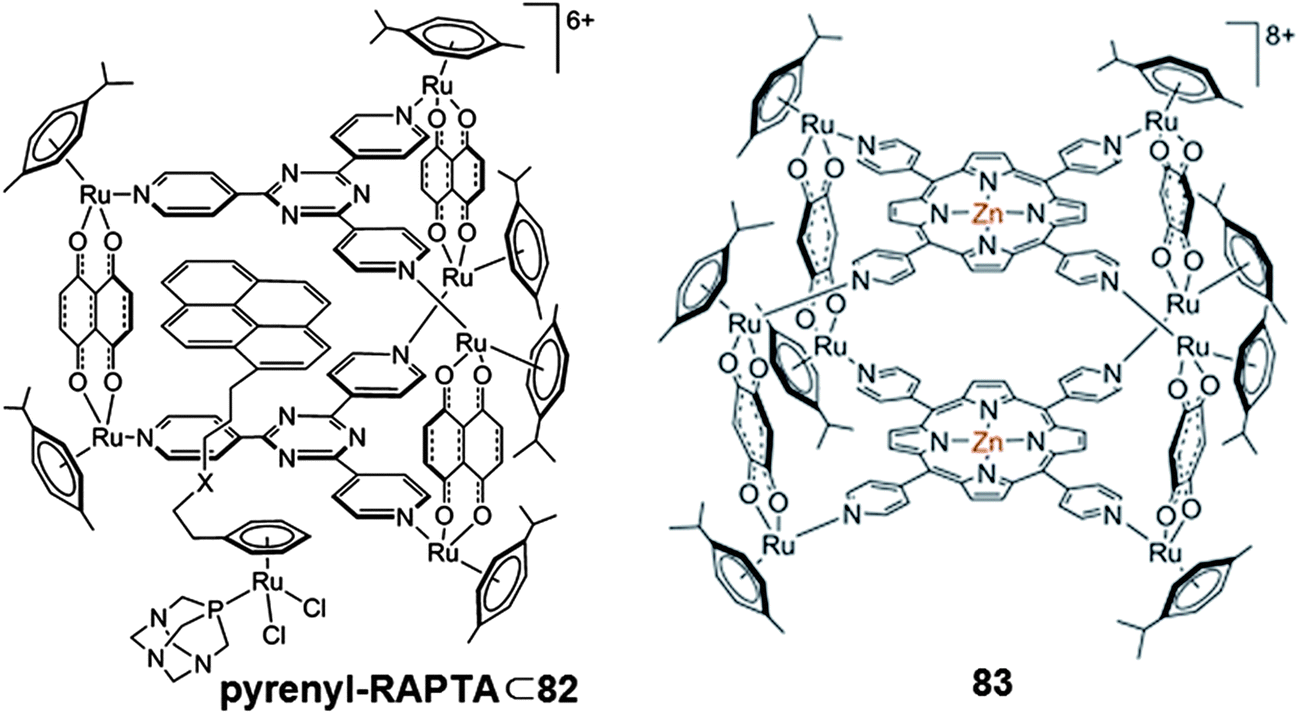 | ||
| Fig. 27 Representative metallacages showing enhanced antiproliferative activity by acting as drug candidates or drug carriers. Adapted and reproduced from ref. 143 and 164. | ||
Dendrimers have emerged as useful scaffolds for visualising the concept of multinuclearity to improve the potency of chemotherapeutic drugs.166–170 The well-defined layered and branched architectures of dendrimers with several terminal groups located at the periphery allows the coordination of various metal centers to the dendrimers. Smith and co-workers explored a series of first and second generation mono-dendrite (N-donor) ruthenium–arene ([{(η6-hexamethylbenzene)RuCl2}8G2], 84 and [{(η6-p-cymene)RuCl2}8G2], 85) metallodendrimers based on second-generation poly(propyleneimine) dendritic scaffolds (G2, Fig. 28).171 The cytotoxicity of these metallodendrimers were examined using the A2780 (human ovarian cancer) cell lines. Though, as compared to cisplatin, these metallodendrimers exhibit modest cytotoxicity with a clear correlation between the cytotoxicity and size of the metallodendrimers. As compared to smaller analogous dendrimers, the octanuclear arene–ruthenium dendrimers 84 and 85 exhibit high activity and ability to readily target cancer cells by entering into the cytoplasm.
 | ||
| Fig. 28 Representative arene–ruthenium metallodendritic complexes based on second generation terminal iminopyridyl dendritic ligands showing cytotoxicity against cancer cells. | ||
Analogous to the above report, cationic and neutral ruthenium arene metallodendrimers with terminal chelating N,O (salicylaldimine) (86) and N,N (iminopyridyl) (87) donor units have also been synthesised and the cytotoxicity of these complexes was examined against A2780 and A2780 cisplatin resistant (human ovarian cancer) cell lines (Fig. 29).172 These arene–ruthenium based metallodendrimers display highly size dependent cytotoxicity. The octanuclear cationic hexamethylbenzene ruthenium based metallodendrimer 87 exhibits the highest activity, compared to the inactive mononuclear complexes. In a recent report on highly branched multinuclear metal dendrimers, Smith and co-workers further explored the extent of size dependent cytotoxicity shown by RAPTA complexes.173 They observed that the 32-armed arene–ruthenium dendrimer (88) exhibits a higher activity than the smaller ones. Replacing chloro- with PTA ligand in these metallodendrimers (88) appears to improve the pharmacological properties of the dendritic systems, leading to a higher cytotoxicity against cancer cells (Fig. 29). Furthermore, the p-cymene based 32-armed metal-dendrimers, 88, exhibit high activity towards the A2780 cancer cells, whereas the hexamethylbenzene based metal-dendrimers show higher activity towards A2780 cisplatin resistant (human ovarian cancer) cell lines. The hexamethylbenzene analogues displayed high cytotoxicity because of their high lipophilicity compared to the p-cymene derivatives or the hexamethylbenzene favour interactions with potential biomolecular targets.
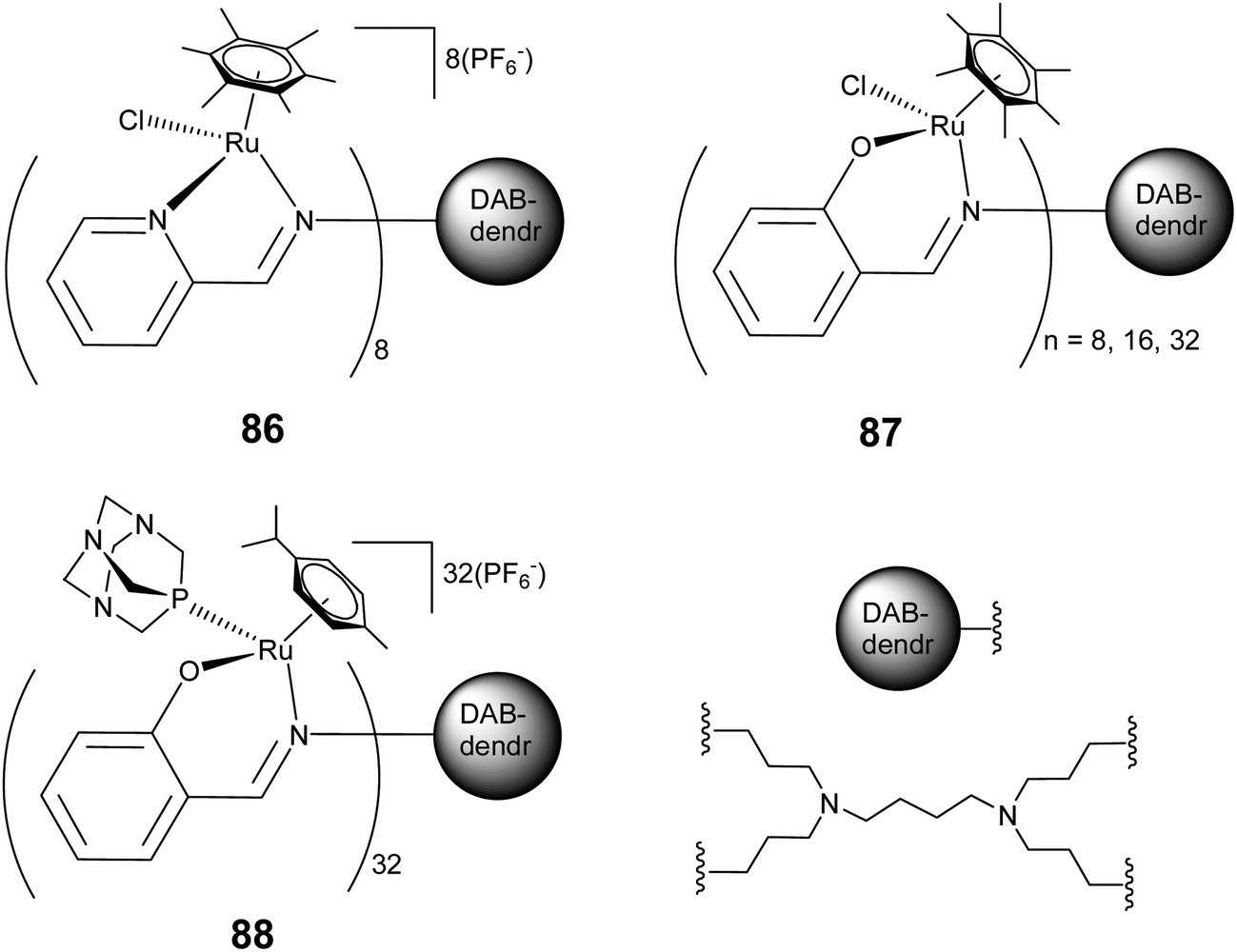 | ||
| Fig. 29 Representative arene–ruthenium metallodendritic complexes based on N,N and N,O donor chelating dendritic ligands showing antiproliferative activity. | ||
7. Conclusions
Arene–ruthenium complexes have emerged as a potential alternative for Pt-based drugs because of their unique structural and electronic behavior. The considerable scope for optimization of the framework of these complexes with a high degree of stability under biological conditions, aqueous solubility, combined lipophilic and hydrophilic character and their affinity towards cancerous cells with considerably low toxicity, are few of the many special features of arene–ruthenium complexes. This review describes the structure and activity relations in arene–ruthenium complexes, with particular emphasis on how the structural features of arene–ruthenium complexes affect their interaction mode with biomolecular targets. The availability of a facile Ru–Cl bond or the presence of a planar pendant moiety, either tagged with arene or linked via a ligand, results in direct bonding or intercalative interaction of arene–ruthenium complexes with DNA. Moreover, kinetically inert half-sandwich ruthenium complexes may also exhibit strong interactions with other biomolecular targets, such as enzymes and act as potential enzyme inhibitors. Furthermore, this review also deals with arene–ruthenium complexes exhibiting photodriven activity for targeted delivery and specific activity against cancer cells. The strategies to combine biologically active organic molecules with an arene–ruthenium moiety are also discussed because these combinations not only have an additive but also a synergistic effect on the overall biological activity. Moreover, the introduction of a multinuclear aspect provides a new strategy for designing new complexes which not only exhibit enhanced activity, but also allow the targeted transport of biologically active molecules to the cellular level (encapsulation of active organic molecules in the cavities of metallacages). Such an intrinsic and controlled behaviour of arene–ruthenium complexes in combination with new strategies, such as, interaction of inert complexes with biomolecules, photodriven activity, metal–drug interactions, along with active multi-nuclear scaffolds, is promising for the development of a unique class of complexes with diverse but targeted biological potentials.Acknowledgements
The authors would like to thank the Editor and anonymous reviewers for their valuable comments and suggestions, which were helpful in improving the article. SKS thanks the Indian Institute of Technology (IIT) Indore, India, and DSP acknowledges the facilities extended by Banaras Hindu University (BHU) Varanasi, and financial support from the Department of Science and Technology (DST), and Council of Scientific and Industrial Research (CSIR), New Delhi, India.References
- W. H. Ang and P. J. Dyson, Eur. J. Inorg. Chem., 2006, 4003–4018 CrossRef CAS.
- C. Gossens, I. Tavernelli and U. Rothlisberger, J. Am. Chem. Soc., 2008, 130, 10921–10928 CrossRef CAS PubMed.
- W. Guo, W. Zheng, Q. Luo, X. Li, Y. Zhao, S. Xiong and F. Wang, Inorg. Chem., 2013, 52, 5328–5338 CrossRef CAS PubMed.
- C. G. Hartinger, M. Groessl, S. M. Meier, A. Casini and P. J. Dyson, Chem. Soc. Rev., 2013, 42, 6186–6199 RSC.
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617–3630 RSC.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- G. Gassr, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef PubMed.
- G. S. Smith and B. Therrien, Dalton Trans., 2011, 40, 10793–10800 RSC.
- A. F. A. Peacock and P. J. Sadler, Chem.–Asian J., 2008, 3, 1890–1899 CrossRef CAS PubMed.
- P. J. Dyson, Chimia, 2007, 61, 698–703 CrossRef CAS.
- M. J. Clarke, Coord. Chem. Rev., 2003, 236, 209–233 CrossRef CAS.
- G. Sava, G. Jaouen, E. A. Hillard and A. Bergamo, Dalton Trans., 2012, 41, 8226–8234 RSC.
- A. Łęczkowska and R. Vilar, Annu. Rep. Prog. Chem., Sect. A, 2013, 109, 299–316 RSC.
- T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013 DOI:10.1021/ar400010v.
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219–5246 RSC.
- G. Sava, A. Bergamo and P. J. Dyson, Dalton Trans., 2011, 40, 9069–9075 RSC.
- K. J. Kilpin and P. J. Dyson, Chem. Sci., 2013, 4, 1410–1419 RSC.
- E. Meggers, Angew. Chem., Int. Ed., 2011, 50, 2442–2448 CrossRef CAS PubMed.
- U. Schatzschneider and N. Metzler-Nolte, Angew. Chem., Int. Ed., 2006, 45, 1504–1507 CrossRef CAS PubMed.
- G. Sava, E. Alessio, A. Bergamo and G. Mestroni, Topics in Biological Inorganic Chemistry, ed. M. J. Clarke and P. J. Sadler, Springer-Verlag, Berlin, 1999, vol. 1, p. 143 Search PubMed.
- A. Bergamo, S. Zorzet, B. Gava, A. Sorc, E. Alessio, E. Iengo and G. Sava, Anti-Cancer Drugs, 2000, 11, 665–672 CrossRef CAS PubMed.
- J. M. Rademaker-Lakhai, D. Van Den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed.
- M. Groessl, C. G. Hartinger, K. Połeć-Pawlak, M. Jarosz, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 1609–1614 CAS.
- M. J. Clarke, Coord. Chem. Rev., 2002, 232, 69–93 CrossRef CAS.
- M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511–2533 CrossRef CAS PubMed.
- A. D. Kelman, M. J. Clake, S. D. Edmonds and H. J. Peresie, J. Clin. Hematol. Oncol., 1977, 7, 274–288 CAS.
- L. D. Dale, J. H. Tocher, T. M. Dyson, D. I. Edwards and D. A. Tocher, Anti-Cancer Drug Des., 1992, 7, 3–14 CAS.
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764–4776 RSC.
- C. S. Allardyce, A. Dorcier, C. Scolaro and P. J. Dyson, Appl. Organomet. Chem., 2005, 19, 1–10 CrossRef CAS.
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC.
- F. Wang, H. Chen, J. A. Parkinson, P. d. S. Murdoch and P. J. Sadler, Inorg. Chem., 2002, 41, 4509–4523 CrossRef CAS PubMed.
- H. Chen, J. A. Parkinson, R. E. Morris and P. J. Sadler, J. Am. Chem. Soc., 2003, 125, 173–186 CrossRef CAS PubMed.
- A. F. A. Peacock, A. Habtemariam, R. Fernández, V. Walland, F. P. A. Fabbiani, S. Parsons, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Am. Chem. Soc., 2006, 128, 1739–1748 CrossRef CAS PubMed.
- T. Bugarcic, O. Nováková, A. Halámiková, L. Zerzánková, O. Vrána, J. Kašpárková, A. Habtemariam, S. Parsons, P. J. Sadler and V. Brabec, J. Med. Chem., 2008, 51, 5310–5319 CrossRef CAS PubMed.
- T. Bugarcic, A. Habtemariam, J. Stepankova, P. Heringova, J. Kasparkova, R. J. Deeth, R. D. L. Johnstone, A. Prescimone, A. Parkin, S. Parsons, V. Brabec and P. J. Sadler, Inorg. Chem., 2008, 47, 11470–11486 CrossRef CAS PubMed.
- P. J. Dyson and G. Sava, Dalton Trans., 2006, 1929–1933 RSC.
- W. H. Ang, C. Scolaro, R. Scopelliti, L. Juillerat-Jeannerat and P. J. Dyson, Inorg. Chem., 2006, 45, 9006–9013 CrossRef CAS PubMed.
- D. V. Deubel and J. K.-C. Lau, Chem. Commun., 2006, 2451–2453 RSC.
- C. Gossens, A. Dorcier, P. J. Dyson and U. Rothlisberger, Organometallics, 2007, 26, 3969–3975 CrossRef CAS.
- A. K. Renfrew, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2010, 49, 2239–2246 CrossRef CAS PubMed.
- A. K. Renfrew, L. Juillerat-Jeanneret and P. J. Dyson, J. Organomet. Chem., 2011, 696, 772–779 CrossRef CAS.
- W. H. Ang, A. Casini, G. Sava and P. J. Dyson, J. Organomet. Chem., 2011, 696, 989–998 CrossRef CAS.
- R. E. Morris, R. E. Aird, P. d. S. Murdoch, H. Chen, J. Cummings, N. D. Hughes, S. Pearsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS PubMed.
- C. S. Allardyce, P. J. Dyson, D. J. Ellis and S. L. Heath, Chem. Commun., 2001, 1396–1397 RSC.
- J. Bravo, S. Bolaño, L. Gonsalvi and M. Peruzzini, Coord. Chem. Rev., 2010, 254, 555–607 CrossRef CAS.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- B. Dutta, C. Scolaro, R. Scopelliti, P. J. Dyson and K. Severin, Organometallics, 2008, 27, 1355–1361 CrossRef CAS.
- C. A. Vock, A. K. Renfrew, R. Scopelliti, L. Juillerat-Jeanneret and P. J. Dyson, Eur. J. Inorg. Chem., 2008, 1661–1671 CrossRef CAS.
- A. K. Renfrew, A. D. Phillips, A. E. Egger, C. G. Hartinger, S. S. Bosquain, A. A. Nazarov, B. K. Keppler, L. Gonsalvi, M. Peruzzini and P. J. Dyson, Organometallics, 2009, 28, 1165–1172 CrossRef CAS.
- A. Dorcier, C. G. Hartinger, R. Scopelliti, R. H. Fish, B. K. Keppler and P. J. Dyson, J. Inorg. Biochem., 2008, 102, 1066–1076 CrossRef CAS PubMed.
- A. Dorcier, P. J. Dyson, C. Goessens, U. Rothlisberger, R. Scopelliti and I. Tavernelli, Organometallics, 2005, 24, 2114–2123 CrossRef CAS.
- K. J. Kilpin, S. M. Cammack, C. M. Clavel and P. J. Dyson, Dalton Trans., 2013, 42, 2008–2014 RSC.
- S. J. Dougan and P. J. Sadler, Chimia, 2007, 61, 704–715 CrossRef CAS.
- C. A. Vock, C. Scolaro, A. D. Phillips, R. Scopelleti, G. Sava and P. J. Dyson, J. Med. Chem., 2006, 49, 5552–5561 CrossRef CAS PubMed.
- S. K. Singh, S. Joshi, A. R. Singh, J. K. Saxena and D. S. Pandey, Inorg. Chem., 2007, 46, 10869–10876 CrossRef CAS PubMed.
- H. M. Chen, J. A. Parkinson, S. Parsons, R. A. Coxall, R. O. Gould and P. J. Sadler, J. Am. Chem. Soc., 2002, 124, 3064–3082 CrossRef CAS PubMed.
- H.-K. Liu, F. Wang, J. A. Parkinson, J. Bella and P. J. Sadler, Chem.–Eur. J., 2006, 12, 6151–6165 CrossRef PubMed.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed.
- F. Wang, H. Chen, S. Parsons, I. D. H. Oswald, J. E. Davidson and P. J. Sadler, Chem.–Eur. J., 2003, 9, 5810–5820 CrossRef CAS PubMed.
- F. Barragán, D. Carrion-Salip, I. Gómez-Pinto, A. González-Cantó, P. J. Sadler, R. de Llorens, V. Moreno, C. González, A. Massaguer and V. Marchán, Bioconjugate Chem., 2012, 23, 1838–1855 CrossRef PubMed.
- H. Wang, N. J. D. Yonker, H. Gao, D. L. Phillips, C. Zhao, L. Ji and Z.-W. Mao, RSC Adv., 2012, 2, 7849–7859 RSC.
- J. Ren, T. C. Jenkins and J. B. Chaires, Biochemistry, 2000, 39, 8439–8447 CrossRef CAS PubMed.
- A. Habtemariam, M. Melchart, R. Fernández, S. Parsons, I. D. H. Oswald, A. Parkin, F. P. A. Fabbiani, J. E. Davidson, A. Dawson, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2006, 49, 6858–6868 CrossRef CAS PubMed.
- F. Wang, J. Xu, K. Wu, S. K. Weidt, C. L. Mackay, P. R. R. Langridge-Smith and P. J. Sadler, Dalton Trans., 2013, 42, 3188–3195 RSC.
- K. D. Camm, A. El-Sokkary, A. L. Gott, P. G. Stockley, T. Belyaeva and P. C. McGowan, Dalton Trans., 2009, 10914–10925 RSC.
- A. D. Phillips, O. Zava, R. Scopelitti, A. A. Nazarov and P. J. Dyson, Organometallics, 2010, 29, 417–427 CrossRef CAS.
- R. Pettinari, C. Pettinari, F. Marchetti, C. M. Clavel, R. Scopelliti and P. J. Dyson, Organometallics, 2013, 32, 309–316 CrossRef CAS.
- A. M. Pizarro and P. J. Sadler, Biochimie, 2009, 91, 1198–1211 CrossRef CAS PubMed.
- M. Melchart, A. Habtemariam, S. Parsons and P. J. Sadler, J. Inorg. Biochem., 2007, 101, 1903–1912 CrossRef CAS PubMed.
- N. Gligorijević, S. Aranđelović, L. Filipović, K. Jakovljević, R. Janković, S. Grgurić-Šipka, I. Ivanović, S. Radulović and Ž. L. Tešić, J. Inorg. Biochem., 2012, 108, 53–61 CrossRef PubMed.
- H.-K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS PubMed.
- O. Novakova, J. Kasparkova, V. Bursova, C. Hofr, M. Vojtiskova, H. Chen, P. J. Sadler and V. Brabec, Chem. Biol., 2005, 12, 121–129 CrossRef CAS PubMed.
- M. Castellano-Castillo, H. Kostrhunova, V. Marini, J. Kasparkova, P. J. Sadler, J. M. Malinge and V. Brabec, JBIC, J. Biol. Inorg. Chem., 2008, 13, 993–999 CrossRef CAS PubMed.
- A. M. Pizarro, A. Habtemariam and P. J. Sadler, Top. Organomet. Chem., 32, 21–56 CrossRef CAS.
- M. Selvakumaran, D. A. Pisarcik, R. Bao, A. T. Yeung and T. C. Hamilton, Cancer Res., 2003, 63, 1311–1316 CAS.
- M. G. Mendoza-Ferri, C. G. Hartinger, A. A. Nazarov, R. E. Eichinger, M. A. Jakupec, K. Severin and B. K. Keppler, Organometallics, 2009, 28, 6260–6265 CrossRef CAS.
- S. J. Lippard, Acc. Chem. Res., 1978, 11, 211–217 CrossRef CAS.
- U. M. Ohndorf, M. A. Rould, Q. He, C. O. Pabo and S. J. Lippard, Nature, 1999, 399, 708–712 CrossRef CAS PubMed.
- Q. Wu, C. Fan, T. Chen, C. Liu, W. Mei, S. Chen, B. Wang, Y. Chen and W. Zheng, Eur. J. Med. Chem., 2013, 63, 57–63 CrossRef CAS PubMed.
- A. Kisova, L. Zerzankova, A. Habtemariam, P. J. Sadler, V. Brabec and J. Kasparkova, Mol. Pharmaceutics, 2011, 8, 949–957 CrossRef CAS PubMed.
- K. J. Kilpin, C. M. Clavel, F. Edafe and P. J. Dyson, Organometallics, 2012, 31, 7031–7039 CrossRef CAS.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC.
- H. Chen, J. A. Parkinson, O. Novakova, J. Bella, F. Y. Wang, A. Dawson, R. Gould, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14623–14628 CrossRef CAS PubMed.
- I. W. McNae, K. Fishburne, A. Habtemariam, T. M. Hunter, M. Melchart, F. Wang, M. D. Walkinshaw and P. J. Sadler, Chem. Commun., 2004, 1786–1787 RSC.
- A. Casini, C. Gabbiani, F. Sorrentino, M. P. Rigobello, A. Bindoli, T. J. Geldbach, A. Marrone, N. Re, C. G. Hartinger, P. J. Dyson and L. Messori, J. Med. Chem., 2008, 51, 6773–6781 CrossRef CAS PubMed.
- L. Oehninger, M. Stefanopoulou, H. Alborzina, J. Schur, S. Ludewig, K. Namikawa, A. Muńoz-Castro, R. W. Köster, K. Baumann, S. Wolfl, W. S. Sheldrick and I. Ott, Dalton Trans., 2013, 42, 1657–1666 RSC.
- J. D. Hayes, J. U. Flanagan and I. R. Jowsey, Annu. Rev. Pharmacol., 2005, 45, 51 CrossRef CAS PubMed.
- W. H. Ang, L. J. Parker, A. D. Luca, J. Juillerat-Jeanneret, C. J. Morton, M. L. Bello, A. W. Parker and P. J. Dyson, Angew. Chem., Int. Ed., 2009, 48, 3854–3857 CrossRef CAS PubMed.
- C. A. Vock, W. H. Ang, C. Scolaro, A. D. Phillips, L. Lagopoulos, L. Juillerat-Jeanneret, G. Sava, R. Scopelleti and P. J. Dyson, J. Med. Chem., 2007, 50, 2166–2175 CrossRef CAS PubMed.
- E. Meggers, G. E. Atilla-Gokcumen, H. Bregman, J. Maksimoska, S. P. Mulcahy, N. Pagano and D. S. Williams, Synlett, 2007, 1177–1189 CrossRef CAS.
- G. E. Atilla-Gokcumen, D. S. Williams, H. Bregman, N. Pagano and E. Meggers, ChemBioChem, 2006, 7, 1443–1450 CrossRef CAS PubMed.
- J. E. Debreczeni, A. X. Bullock, G. E. Atilla-Gokcumen, D. S. Williams, H. Bregman, S. Knopp and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 1580–1585 CrossRef CAS PubMed.
- J. Maksimoska, L. Feng, C. Yi, J. Kissil, R. Marmorstein and E. Meggers, J. Am. Chem. Soc., 2008, 130, 15764–15765 CrossRef CAS PubMed.
- P. Xie, D. S. Williams, G. E. Atilla-Gokcumen, L. Milk, M. Xiao, K. S. M. Smalley, M. Herlyn, E. Meggers and R. Marmorstein, ACS Chem. Biol., 2008, 3, 305–316 CrossRef CAS PubMed.
- S. Blanck, T. Cruchter, A. Vultur, R. Riedel, K. Harms, M. Herlyn and E. Meggers, Organometallics, 2011, 30, 4598–4606 CrossRef CAS PubMed.
- P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC.
- S. J. Dougan, A. Habtemariam, S. E. McHale, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11628–11633 CrossRef CAS PubMed.
- I. Romero-Canelon, A. M. Pizarro, A. Habtemariam and P. J. Sadler, Metallomics, 2012, 4, 1271–1279 RSC.
- I. Romero-Canelon, L. Salassa and P. J. Sadler, J. Med. Chem., 2013, 56, 1291–1300 CrossRef CAS PubMed.
- S. J. Dougan, M. Melchart, A. Habtemariam, S. Parsons and P. J. Sadler, Inorg. Chem., 2006, 45, 10882–10894 CrossRef CAS PubMed.
- F. Giannini, G. Suss-Fink and J. Furrer, Inorg. Chem., 2011, 50, 10552–10554 CrossRef CAS PubMed.
- W. Ying, Antioxid. Redox Signaling, 2008, 10, 179–206 CrossRef CAS PubMed.
- P. Belenky, K. L. Bogan and C. Brenner, Trends Biochem. Sci., 2007, 32, 12–19 CrossRef CAS PubMed.
- S.-J. Lin and L. Guarente, Curr. Opin. Cell Biol., 2003, 15, 241–246 CrossRef CAS PubMed.
- Y. Yan, M. Melchart, A. Habtemariam, A. Peacock and P. J. Sadler, JBIC, J. Biol. Inorg. Chem., 2006, 11, 483–488 CrossRef CAS PubMed.
- S. Betanzos-Lara, Z. Liu, A. Habtemariam, A. M. Pizarro, B. Qamar and P. J. Sadler, Angew. Chem., Int. Ed., 2012, 51, 3897–3900 CrossRef CAS PubMed.
- U. Schatzschneider, Eur. J. Inorg. Chem., 2010, 1451–1467 CrossRef CAS.
- S. Betanzos-Lara, L. Salassa, A. Habtemariam and P. J. Sadler, Chem. Commun., 2009, 6622–6624 RSC.
- F. Barragán, P. López-Senín, L. Salassa, S. Betanzos-Lara, A. Habtemariam, V. Moreno, P. J. Sadler and V. Marchán, J. Am. Chem. Soc., 2011, 133, 14098–14108 CrossRef PubMed.
- S. Betanzos-Lara, L. Salassa, A. Habtemariam, O. Novakova, A. M. Pizarro, G. J. Clarkson, B. Liskova, V. Brabec and P. J. Sadler, Organometallics, 2012, 31, 3466–3479 CrossRef CAS.
- Y. Chen, W. Lei, G. Jiang, Q. Zhou, Y. Hou, C. Li, B. Zhang and X. Wang, Dalton Trans., 2013, 42, 5924–5931 RSC.
- S. W. Magennis, A. Habtemariam, O. Novákova, J. B. Henry, S. Meier, S. Parsons, I. D. H. Oswald, V. Brabec and P. J. Sadler, Inorg. Chem., 2007, 46, 5059–5068 CrossRef CAS PubMed.
- F. Schmitt, P. Govindaswamy, G. Suss-Fink, W. H. Ang, P. J. Dyson, L. Juillerat-Jeanneret and B. Therrien, J. Med. Chem., 2008, 51, 1811–1816 CrossRef CAS PubMed.
- M. Pernot, T. Bastogne, N. P. E. Barry, B. Therrien, G. Koellensperger, S. Hann, V. Reshetov and M. Barberi-Heyob, J. Photochem. Photobiol., B, 2012, 117, 80–89 CrossRef CAS PubMed.
- F. Schmitt, P. Govindaswamy, O. Zava, G. Suss-Fink, L. Juillerat-Jeanneret and B. Therrien, JBIC, J. Biol. Inorg. Chem., 2009, 14, 101–109 CrossRef CAS PubMed.
- F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeaneret, G. Suss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754–757 CrossRef CAS PubMed.
- W. F. Schmid, S. Zorbas-Seifried, R. O. John, V. B. Arion, M. A. Jakupec, A. Roller, M. Galanski, I. Chiorescu, H. Zorbas and B. K. Keppler, Inorg. Chem., 2007, 46, 3645–3656 CrossRef CAS PubMed.
- W. F. Schmid, R. O. John, V. B. Arion, M. A. Jakupec and B. K. Keppler, Organometallics, 2007, 26, 6643–6652 CrossRef CAS.
- W. F. Schmid, R. O. John, G. Mühlgassner, P. Heffeter, M. A. Jakupec, M. Galanski, W. Berger, V. B. Arion and B. K. Keppler, J. Med. Chem., 2007, 50, 6343–6355 CrossRef CAS PubMed.
- W. Zheng, Q. Luo, Y. Lin, Y. Zhao, X. Wang, Z. Du, X. Hao, Y. Yu, S. Lü, L. Ji, X. Li, L. Yang and F. Wang, Chem. Commun., 2013, 49, 10224–10226 RSC.
- C. S. K. Rajapakse, A. Martínez, B. Naoulou, A. A. Jarzecki, L. Suárez, C. Deregnaucourt, V. Sinou, J. Schrével, E. Musi, G. Ambrosini, G. K. Schwartz and R. A. Sánchez-Delgado, Inorg. Chem., 2009, 48, 1122–1131 CrossRef CAS PubMed.
- W. Ginzinger, G. Mühlgassner, V. B. Arion, M. A. Jakupec, A. Roller, M. Galanski, M. Reithofer, W. Berger and B. K. Keppler, J. Med. Chem., 2012, 55, 3398–3413 CrossRef CAS PubMed.
- A. Martínez, T. Carreon, E. Iniguez, A. Anzellotti, A. Sánchez, M. Tyan, A. Sattler, L. Herrera, R. A. Maldonado and R. A. Sánchez-Delgado, J. Med. Chem., 2012, 55, 3867–3877 CrossRef PubMed.
- A. A. Nazarov, D. Gardini, M. Baquié, L. Juillerat-Jeanneret, T. P. Serkova, E. P. Shevtsova, R. Scopelliti and P. J. Dyson, Dalton Trans., 2013, 42, 2347–2350 RSC.
- I. Turel, Coord. Chem. Rev., 2002, 232, 27–47 CrossRef CAS.
- M. E. Katsarou, E. K. Efthimiadou, G. Psomas, A. Karaliota and D. Vourloumis, J. Med. Chem., 2008, 51, 470–478 CrossRef CAS PubMed.
- I. Turel, J. Kljun, F. Perdih, E. Morozova, V. Bakulev, N. Kasyanenko, J. A. W. Byl and N. Osheroff, Inorg. Chem., 2010, 49, 10750–10752 CrossRef CAS PubMed.
- J. Kljun, A. K. Bytzek, W. Kandioller, C. Bartel, M. A. Jakupec, C. G. Hartinger, B. K. Keppler and I. Turel, Organometallics, 2011, 30, 2506–2512 CrossRef CAS PubMed.
- R. Hudej, J. Kljun, W. Kandioller, U. Repnik, B. Turk, C. G. Hartinger, B. K. Keppler, D. Miklavčič and I. Turel, Organometallics, 2012, 31, 5867–5874 CrossRef CAS.
- L. K. Filak, G. Mühlgassner, F. Bacher, A. Roller, M. Galanski, M. A. Jakupec, B. K. Keppler and V. B. Arion, Organometallics, 2011, 30, 273–283 CrossRef CAS PubMed.
- L. K. Filak, S. Goschl, S. Hackl, M. A. Jakupec and V. B. Arion, Inorg. Chem., 2012, 393, 252–260 CAS.
- L. K. Filak, S. Goschl, P. Heffeter, K. G. Samper, A. E. Egger, M. A. Jakupec, B. K. Keppler, W. Berger and V. B. Arion, Organometallics, 2013, 32, 903–914 CrossRef CAS PubMed.
- M. Grazul and E. Budzisz, Coord. Chem. Rev., 2009, 253, 2588–2598 CrossRef CAS.
- A. Kurzwernhart, W. Kandioller, C. Bartel, S. Bächler, R. Trondl, G. Mühlgassner, M. A. Jakupec, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2012, 48, 4839–4841 RSC.
- A. Kurzwernhart, W. Kandioller, S. Bachler, C. Bartel, S. Martic, M. Buczkowska, G. Muhlgassner, M. A. Jakupec, H.-B. Kraatz, P. J. Bendarski, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, J. Med. Chem., 2012, 55, 10512–10522 CrossRef CAS PubMed.
- A. Kurzwernhart, W. Kandioller, E. A. Enyedy, M. Novak, M. A. Jakupec, B. K. Keppler and C. G. Hartinger, Dalton Trans., 2013, 42, 6193–6202 RSC.
- W. Kandioller, E. Balsano, S. M. Meier, U. Jungwirth, S. Göschl, A. Roller, M. A. Jakupec, W. Berger, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2013, 49, 3348–3350 RSC.
- F. Caruso, M. Rossi, A. Benson, C. Opazo, D. Freedman, E. Monti, M. B. Gariboldi, J. Shaulky, F. Marchetti, R. Pettinari and C. Pettinari, J. Med. Chem., 2012, 55, 1072–1081 CrossRef CAS PubMed.
- F. Yang, P. L. G. P. Lim, A. N. Begum, O. J. Ubeda, M. R. Simmons, S. S. Ambegaokar, P. Chen, R. Kayed, C. G. Glabe, S. A. Frautschy and G. M. Cole, J. Biol. Chem., 2004, 280, 5892–5901 CrossRef PubMed.
- B. B. Aggarwal, C. Sundaram, N. Malani and H. Ichikawa, Adv. Exp. Med. Biol., 595, 1–75 CrossRef.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- N. P. E. Barry, N. H. A. Karim, R. Vilar and B. Therrien, Dalton Trans., 2009, 10717–10719 RSC.
- N. P. E. Barry, O. Zava, J. Furrer, P. J. Dyson and B. Therrien, Dalton Trans., 2010, 39, 5272–5277 RSC.
- M.-G. Mendoza-Ferri, C. G. Hartinger, R. E. Eichinger, N. Stolyarova, K. Severin, M. A. Jakupec, A. A. Nazarov and B. K. Keppler, Organometallics, 2008, 27, 2405–2407 CrossRef CAS.
- M.-G. Mendoza-Ferri, C. G. Hartinger, M. A. Mendoza, M. Groessl, A. E. Egger, R. E. Eichinger, J. B. Mangrum, N. P. Farrell, M. Maruszak, P. J. Bednarski, F. Klein, M. A. Jakupec, A. A. Nazarov and B. K. Keppler, J. Med. Chem., 2009, 52, 916–925 CrossRef CAS PubMed.
- M. Auzias, B. Therrien, G. Süss-Fink, P. Štěpnička, W. H. Ang and P. J. Dyson, Inorg. Chem., 2008, 47, 578–583 CrossRef CAS PubMed.
- M. Gras, B. Therrien, G. Süss-Fink, O. Zava and P. J. Dyson, Dalton Trans., 2010, 39, 10305–10313 RSC.
- W. H. Ang, Z. Grote, R. Scopelliti, L. Juillerat-Jeanneret, K. Severin and P. J. Dyson, J. Organomet. Chem., 2009, 694, 968–972 CrossRef CAS.
- J. Mattsson, P. Govindaswamy, A. K. Renfrew, P. J. Dyson, P. Štĕpnička, G. Süss-Fink and B. Therrien, Organometallics, 2009, 28, 4350–4357 CrossRef CAS.
- N. P. E. Barry, F. Edafe and B. Therrien, Dalton Trans., 2011, 40, 7172–7180 RSC.
- F. Linares, E. Q. Procopio, M. A. Galindo, M. A. Romero, J. A. R. Navarro and E. Barea, CrystEngComm, 2010, 12, 2343–2346 RSC.
- A. Mishra, H. Jung, J. W. Park, H. K. Kim, H. Kim, P. J. Stang and K.-W. Chi, Organometallics, 2012, 31, 3519–3526 CrossRef CAS PubMed.
- V. Vajpayee, Y. H. Song, Y. J. Jung, S. C. Kang, H. Kim, I. S. Kim, M. Wang, T. R. Cook, P. J. Stang and K.-W. Chi, Dalton Trans., 2012, 41, 3046–3052 RSC.
- V. Vajpayee, S. Lee, S.-H. Kim, S. C. Kang, T. R. Cook, H. Kim, D. W. Kim, S. Verma, M. S. Lah, I. S. Kim, M. Wang, P. J. Stang and K.-W. Chi, Dalton Trans., 2013, 42, 466–475 RSC.
- V. Vajpayee, Y. H. Song, Y. J. Yang, S. C. Kang, H. Kim, I. S. Kim, M. Wang, P. J. Stang and K.-W. Chi, Organometallics, 2011, 30, 3242–3245 CrossRef CAS PubMed.
- V. Vajpayee, S. mi Lee, J. W. Park, A. Dubey, H. Kim, T. R. Cook, P. J. Stang and K.-W. Chi, Organometallics, 2013, 32, 1563–1566 CrossRef CAS PubMed.
- V. Vajpayee, Y. J. Yang, S. C. Kang, H. Kim, I. S. Kim, M. Wang, P. J. Stang and K.-W. Chi, Chem. Commun., 2011, 47, 5184–5186 RSC.
- J. Mattsson, P. Govindaswamy, J. Furrer, Y. Sei, K. Yamaguchi, G. Süss-Fink and B. Therrien, Organometallics, 2008, 27, 4346–4356 CrossRef CAS.
- B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS PubMed.
- J. W. Yi, N. P. E. Barry, M. A. Furrer, O. Zava, P. J. Dyson, B. Therrien and B. H. Kim, Bioconjugate Chem., 2012, 23, 461–471 CrossRef CAS PubMed.
- K. M. Guckian, B. A. Schweitzer, R. X. F. Ren, C. J. Sheils, D. C. Tahmassebi and E. T. Kool, J. Am. Chem. Soc., 2000, 122, 2213–2222 CrossRef CAS PubMed.
- L. Hernandez-Folgado, D. Baretić, I. Piantanida, M. Marjanović, M. Kralj, T. Rehm and C. Schmuck, Chem.–Eur. J., 2010, 16, 3036–3056 CrossRef CAS PubMed.
- M. A. Furrer, F. Schmitt, M. Wiederkehr, L. Juillerat-Jeanneret and B. Therrien, Dalton Trans., 2012, 41, 7201–7211 RSC.
- N. P. E. Barry, O. Zava, P. J. Dyson and B. Therrien, Aust. J. Chem., 2010, 63, 1529–1537 CrossRef CAS.
- P. Govender, B. Therrien and G. S. Smith, Eur. J. Inorg. Chem., 2012, 2853–2862 CrossRef CAS.
- C. Billecke, S. Finniss, L. Tahash, C. Miller, T. Mikkelsen, N. P. Farrell and O. Bogler, Neuro-Oncology, 2006, 8, 215–226 CrossRef CAS PubMed.
- B. A. Jansen, J. van der Zwan, J. Reedijk, H. den Dulk and J. Brouwer, Eur. J. Inorg. Chem., 1999, 1429–1433 CrossRef CAS.
- T. Kapp, A. Dullin and R. Gust, J. Med. Chem., 2006, 49, 1182–1190 CrossRef CAS PubMed.
- T. Kapp, A. Dullin and R. Gust, Bioconjugate Chem., 2010, 21, 328–337 CrossRef CAS PubMed.
- P. Govender, N. C. Antonels, J. Mattsson, A. K. Renfrew, P. J. Dyson, J. R. Moss, B. Therrien and G. S. Smith, J. Organomet. Chem., 2009, 694, 3470–3476 CrossRef CAS.
- P. Govender, A. K. Renfrew, C. M. Clavel, P. J. Dyson, B. Therrien and G. S. Smith, Dalton Trans., 2011, 40, 1158–1167 RSC.
- P. Govender, L. C. Sudding, C. M. Vlavel, P. J. Dyson, B. Therrien and G. S. Smith, Dalton Trans., 2013, 42, 1267–1277 RSC.
| This journal is © The Royal Society of Chemistry 2014 |