Structure and thermal properties of composites with RE-borohydrides (RE = La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Er, Yb or Lu) and LiBH4†
Jørn Eirik
Olsen
a,
Christoph
Frommen
a,
Torben R.
Jensen
b,
Marit D.
Riktor
a,
Magnus H.
Sørby
a and
Bjørn C.
Hauback
*a
aInstitute for Energy Technology, Physics Department, P.O. Box 40, NO-2027 Kjeller, Norway. E-mail: bjorn.hauback@ife.no; Fax: +47 63 81 09 20; Tel: +47 63 80 60 78
bCenter for Materials Crystallography (CMC), Interdisciplinary Nanoscience Center (iNANO) and Department of Chemistry, Aarhus University, Langelandsgade 140, DK-8000 C, Aarhus, Denmark
First published on 31st October 2013
Abstract
RECl3 (RE = La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Er, Yb, Lu) and LiBH4 ball-milled in the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 resulted in formation of numerous rare-earth metal (RE) borohydrides and LiCl in mixtures with excess LiBH4. The reaction products were characterized by powder X-ray diffraction and infrared spectroscopy, and their hydrogen sorption properties were investigated by thermogravimetric analysis combined with differential scanning calorimetry and in situ synchrotron radiation powder X-ray diffraction. La, Ce, Pr and Nd form LiRE(BH4)3Cl compounds which crystallize in the cubic space group I
:6 resulted in formation of numerous rare-earth metal (RE) borohydrides and LiCl in mixtures with excess LiBH4. The reaction products were characterized by powder X-ray diffraction and infrared spectroscopy, and their hydrogen sorption properties were investigated by thermogravimetric analysis combined with differential scanning calorimetry and in situ synchrotron radiation powder X-ray diffraction. La, Ce, Pr and Nd form LiRE(BH4)3Cl compounds which crystallize in the cubic space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m. Sm, Gd, Tb, Er and Yb form RE(BH4)3 phases crystallizing in the cubic space group Pa
3m. Sm, Gd, Tb, Er and Yb form RE(BH4)3 phases crystallizing in the cubic space group Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , with a possible polymorphic transition to a higher symmetry space group, Pmm. The smaller RE-elements Yb and Lu form tetrahedral [RE(BH4)4]− anionic complexes stabilized by Li+ cations crystallizing in the tetragonal space group P2c. Additionally Sm and Gd also exhibit a transition to the LiRE(BH4)3Cl phases observed for the largest lanthanides. LiSm(BH4)3Cl is reduced to Sm(BH4)2 upon heating, which takes a previously unreported orthorhombic structure in space group Pbcn. Eu did not form any of these RE-borohydride compounds, but infrared spectroscopy indicates formation of a new borohydride phase. Hydrogen desorption from the composite mixtures starts around 200 °C. Rehydrogenation of the La-system under p(H2) = 10 MPa at 300 °C showed a partial reversibility, with 18% recovery of the original hydrogen capacity, which increased to 36% at 415 °C. Rehydrogenation under p(H2) = 10 MPa at 400 °C showed a partial reversibility of 25% in the Er-system. Formation of REH2 during thermal decomposition destabilizes the remaining LiBH4 in the composites, thus releasing hydrogen at lower temperatures than for pure LiBH4.
, with a possible polymorphic transition to a higher symmetry space group, Pmm. The smaller RE-elements Yb and Lu form tetrahedral [RE(BH4)4]− anionic complexes stabilized by Li+ cations crystallizing in the tetragonal space group P2c. Additionally Sm and Gd also exhibit a transition to the LiRE(BH4)3Cl phases observed for the largest lanthanides. LiSm(BH4)3Cl is reduced to Sm(BH4)2 upon heating, which takes a previously unreported orthorhombic structure in space group Pbcn. Eu did not form any of these RE-borohydride compounds, but infrared spectroscopy indicates formation of a new borohydride phase. Hydrogen desorption from the composite mixtures starts around 200 °C. Rehydrogenation of the La-system under p(H2) = 10 MPa at 300 °C showed a partial reversibility, with 18% recovery of the original hydrogen capacity, which increased to 36% at 415 °C. Rehydrogenation under p(H2) = 10 MPa at 400 °C showed a partial reversibility of 25% in the Er-system. Formation of REH2 during thermal decomposition destabilizes the remaining LiBH4 in the composites, thus releasing hydrogen at lower temperatures than for pure LiBH4.
Introduction
Metal borohydrides have been extensively investigated over the last few years as potential hydrogen storage materials for mobile applications1–5 due to their high volumetric and gravimetric hydrogen capacities, e.g. 18.5 wt% hydrogen in LiBH4.6 Unfortunately the pure lightweight alkali metal borohydrides are proven unsuitable for practical hydrogen storage, due to high desorption temperatures and limited reversibility at moderate conditions.7,8 DFT calculations have predicted favorable thermodynamic properties of the alkaline-earth metal borohydrides Mg(BH4)2 and Ca(BH4)2, but they are plagued by slow kinetics and limited reversibility at moderate conditions.9–14Transition metal borohydrides based on zinc or cadmium, e.g. LiZn2(BH4)5 or Cd(BH4)2, are too unstable and decompose to diborane (B2H6) and hydrogen at T ∼100 °C, and manganese borohydride may also release some diborane at slightly higher temperatures.5,15,16 The stability of metal borohydrides in terms of their decomposition temperatures has been found to inversely correlate with the Pauling electronegativity (χP) of the metal cation.17–19 A similar correlation has been suggested in mixed metal borohydrides, using the averaged χP of the two metals.20,21 However, in mixed metal borohydrides containing complex anions the desorption temperature is dependent on the χP of the metal that coordinates to BH4,3 which indicates a key role of the complex anion in the structural stability of mixed metal borohydrides.
Recently interest has been directed to the synthesis and properties of transition metal- and rare-earth (RE) borohydrides. Mechanochemical milling of RECl3 with LiBH4 has resulted in four different structure types of RE-borohydride phases: LiRE(BH4)4 (RE = Sc,22 Yb23), α-RE(BH4)3 (RE = Y,24 Gd,24 Dy,24 Yb23), β-RE(BH4)3 (RE = Y,25–27 Yb23) and LiRE(BH4)3Cl (RE = La,28 Ce,29,30 Gd28). The LiRE(BH4)4 phases take a primitive tetragonal structure in the space group P2c, and are composed of distorted tetrahedral RE(BH4)4− anions stabilized by Li+ cations. The RE(BH4)3 compounds exhibit polymorphism where α-RE(BH4)3 takes the cubic space group Pa with a distorted octahedral arrangement of BH4− units around RE, while β-RE(BH4)3 has an ideal octahedral arrangement with the cubic space groups Pmm or Fmc. The relationship between the two structure models is discussed in ref. 23. The LiRE(BH4)3Cl compounds take a body centered cubic structure in the space group I3m where each RE is octahedrally coordinated by 3 BH4− units and 3 Cl atoms, while the RE and Cl atoms in the unit cell form a distorted RE4–Cl4 cube.
The RE–BH4 compounds obtained by mechanochemical milling of RECl3 + 3LiBH4 decompose between 200 and 300 °C,25,28–32 which is considerably lower than the decomposition temperature of LiBH4 at 380 °C.33 Recently Gennari et al.34 showed that composites of RECl3 + 6LiBH4 (RE = Ce, Gd) form rare-earth hydrides during thermal decomposition which in turn destabilize the remaining LiBH4. Partial reversibility (40%) was obtained during rehydrogenation at 6 MPa and 400 °C. Furthermore decomposition against 0.5 MPa H2 backpressure altered the decomposition pathway in comparison to decomposition under vacuum, promoting the formation of CeB6 and GdB4, respectively.
In addition to their hydrogen storage properties the Li containing RE borohydrides LiRE(BH4)3Cl are also of interest due to their high Li-ion conductivity at room temperature (RT),28,30 which could make them attractive as solid state electrolyte in Li-ion batteries. The high Li ion conductivity may be due to correlated motion of Li ions, diffusion and reorientational motion of BH4 as found by solid state NMR of LiLa(BH4)3Cl.35
In this paper, we present a wide range of novel rare earth borohydrides based on RE = La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Er, Yb and Lu and discover trends in crystal chemistry and properties. Moreover, the thermal decomposition behaviours in composites with LiBH4 are reported.
Experimental
Sample preparation
LiBH4 (Sigma-Aldrich, >95%) and RECl3 (RE = La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Er, Yb or Lu) (Sigma-Aldrich, 99.99%) in 6:1 molar ratio were milled in a Fritsch Pulverisette 7 at 400 rpm with milling time of 5 h. Stainless steel vial and balls were used with a ball-to-powder ratio of 40:1.
All sample handling was performed under inert Ar-atmosphere in an MBraun Unilab glove box fitted with a recirculation system and oxygen/humidity sensors. Oxygen and water levels were kept below 1 ppm during all operations.
Additional annealing at 100 °C for 50 h was done within boron glass capillaries to improve crystallinity for powder diffraction measurements, except for the in situ measurements.
Powder X-ray diffraction (PXD)
Patterns were collected in transmission mode using CuKα radiation (λ = 1.5418 Å) in a Bruker AXS D8 Advance as described in ref. 23 but with the 2θ range 10 to 60°.Infrared (IR) spectroscopy
Spectra were collected on a single-reflection BRUKER ALPHA-Platinum ATR (attenuated total refection) instrument with a diamond crystal, placed inside the glove box. The measurements were performed in the 4000–400 cm−1 range, with 2 cm−1 resolution. The spectra were averaged over 32 scans and background subtracted.Thermogravimetric analysis/differential scanning calorimetry (TG/DSC)
Measurements were conducted in a Netzsch STA 449 F3 Jupiter instrument as described in ref. 23 with sample masses of approximately 10 mg and 5 °C min−1 heating rate up to 400 °C. The desorbed gases were analysed with a MKS Microvision-IP residual gas analyzer (RGA) by mass spectroscopy (MS).Synchrotron radiation powder X-ray diffraction (SR-PXD)
Measurements were performed at the Swiss-Norwegian Beam Line (SNBL), BM01A at ESRF, Grenoble, France. The diffraction data were collected with a PILATUS2M detector at the diffractometer PILATUS@SNBL, using the software Pylatus developed at SNBL. RT measurements were performed with a sample-to-detector distance of 310 mm and a wavelength of 0.69660 Å. The samples were contained in boron glass capillaries (0.5 mm diameter) filled and sealed under Ar atmosphere. Exposure times of 30 or 40 s were used, and the capillary was rotated 10° during exposure to improve the powder averaging.In situ SR-PXD measurements were performed with an in-house build gas rig and sample holder, with the sample contained within a sapphire tube (inner diameter 0.8 mm, outer diameter 1.15 mm). Sample-to-detector distances of 310 or 410 mm and λ = 0.694118 Å were used. The samples were thermally decomposed either under dynamic vacuum or 0.5 MPa hydrogen backpressure from RT to 300 or 400 °C with a heating rate of 3.75 or 5.00 °C min−1. Subsequent hydrogenation was performed in some cases under 10 MPa hydrogen pressure at the maximum temperature. The temperature was kept at 300 °C for 30 minutes before cooling down to RT in 30 minutes.
The two-dimensional data from the PILATUS2M area-detector were integrated as described in ref. 23.
Structure solution and refinement
Powder indexing and structure solution were performed with the programs TREOR36 and FOX,37 respectively. Structural refinement using the Rietveld method was performed using the GSAS software package, with the graphical interface EXPGUI.38,39 The BH4− tetrahedral units were treated as semi-rigid bodies by restraints on the B–H and H–H distances, as 1.2(±0.1) Å and 1.95(±0.1) Å, respectively. The Bragg peak profiles were described by a pseudo-Voigt function with up to 6 free parameters and Shifted Chebyschev functions with 20 parameters were used to fit the background. The standard deviations of the intensity values for the SR-PXD patterns were included in the data file for GSAS by the program Powder4.Results/discussion
Structural characterization
RE: La, Ce, Pr and Nd. The dominant set of Bragg peaks observed for the annealed samples with RE = La, Ce, Pr and Nd could be indexed based on a cubic unit cell with space group I
3m, consistent with the reported structure of LiRE(BH4)3Cl, RE = La, Ce, Gd.28–30 In addition, all data sets show peaks from LiCl and excess LiBH4, while there are peaks from RECl3. The diffraction pattern for the Nd-sample also include strong peaks corresponding to NdOCl, which was found to be an impurity in the starting material NdCl3. Structure refinement of the LiRE(BH4)3Cl phases at RT using the Rietveld method was performed based on the SR-PXD data. The result is shown for RE = La in Fig. 1 and similar fits were obtained for RE = Ce, Pr and Nd. The refined unit cell dimensions are given in Table 1 with refined crystallographic data and selected RE–B and RE–Cl distances in the ESI (Tables S1 and S3–S6†). The unit cell dimensions and the RE–Cl distances vary in agreement with the lanthanide contraction,40 while the RE–B distances are almost constant at approx. 2.72 Å.
 | ||
| Fig. 1 Rietveld refinement plot from SR-PXD patterns at RT for the different ball-milled and annealed samples of LaCl3 + 6 LiBH4 showing experimental (circles) and calculated (red line) patterns, and a difference plot (below). Tick marks show positions of Bragg peaks from LiLa(BH4)3Cl (bottom), LiCl, LiBH4, LaCl3 (top), Rwp = 2.58%. | ||
| Element | Ionic radii, Å | α-RE(BH4)3, Å | β-RE(BH4)3, Å | LiRE(BH4)3Cl, Å |
|---|---|---|---|---|
| a Ref. 24. b Measured at the high resolution SR-PXD instrument at BM01B, SNBL, ESRF, Grenoble, France. c Ref. 23. | ||||
| La | 1.032 | — | — | 11.6853(1) |
| Ce | 1.01 | — | — | 11.6243(1) |
| Pr | 0.99 | — | — | 11.5784(1) |
| Nd | 0.983 | — | — | 11.5480(2) |
| Sm | 0.958 | 11.0959(3) | 5.6406(1) | — |
| Eu | 0.947 | — | — | — |
| Gd | 0.935 | 11.0255(8) | — | — |
| Tb | 0.923 | 10.9125(2)b | — | — |
| Dy | 0.912 | 10.885a | — | — |
| Ho | — | — | — | |
| Er | 0.890 | 10.7790(7)b | 5.4790(1) | — |
| Tm | 0.880 | — | — | — |
| Yb | 0.868 | 10.70715(15)c | 5.44223(3)c | — |
RE = Sm, Eu, Gd, Tb, Er, Yb and Lu. For the Sm sample, Bragg peaks from two phases were present in addition to LiCl and LiBH4. They were indexed with two primitive cubic unit cells with dimensions a = 11.10 Å and a = 5.64 Å and extinction rules according to the space groups Pa
and Pmm respectively. These phases are similar to those reported for α-RE(BH4)3 (ref. 23 and 24) and β-RE(BH4)3,23,25,26 and therefore named α-Sm(BH4)3 and β-Sm(BH4)3. The PXD pattern for the Eu-sample only included weak peaks, which could not be identified as any known Eu-containing phases, and no other phases than LiCl were readily determined. In the Gd sample a set of peaks could be indexed on a primitive cubic unit cell, consistent with the reported α-Gd(BH4)3 structure.24 The PXD pattern of the Tb sample included peaks similar to α-RE(BH4)3, and thus the phase was named α-Tb(BH4)3. In the Er sample, a set of peaks which could be indexed by a primitive cubic unit cell was observed, similar to the β-RE(BH4)3, therefore this phase was named β-Er(BH4)3. The annealed Yb-sample did not contain Bragg peaks from the reported trivalent borohydrides, rather a divalent decomposition product ortho-Yb(BH4)2, consistent with the previously reported decomposition around 100 °C for the trivalent borohydride.23 The PXD pattern of the Lu-sample included a set of peaks similar to the reported tetragonal phases LiRE(BH4)4, and thereby identified as LiLu(BH4)4.
We have also been able to synthesize α-Er(BH4)3 from ball-milling 3LiBH4 + ErCl3. This means that samples of RECl3 with RE = Sm, Gd, Tb, Y,24 Dy,24 Er and Yb23 ball milled with LiBH4 may all contain RE borohydride with the α-RE(BH4)3 type structure (Pa). In addition the β-RE(BH4)3 type structures (Pmm or Fmc) are observed at RT for Sm, Y,25,26 Er and Yb.23 For the smaller RE elements Yb and Lu the LiRE(BH4)4 type structure (P2c) are also formed. A schematic overview of borohydride phases observed in the composites can be found in Table 2.
Table 1 shows the refined unit cell dimensions for the RE–borohydrides from SR-PXD data in the present work. Previously reported unit cell dimensions for similar compounds are included for comparison. The phases are listed in descending order of ionic radii of the RE element. There is a linear correlation between the unit cell dimensions and the ionic radii of the RE cation in the series of isostructural compounds.
Infrared spectroscopy
IR spectra for all 6:1 samples (Fig. S1 in ESI†) were compared to the spectrum of pure LiBH4, and they were found to include modes from LiBH4. IR spectra for the ball milled RECl3 + 6 LiBH4 samples (RE = La, Eu, Er, Lu) and pure LiBH4 are plotted in Fig. 2. A strong additional band at 1176 cm−1 with a shoulder at 1201 cm−1 in the B–H bending region, was attributed to LiLa(BH4)3Cl. The stretching modes assigned to LiLa(BH4)3Cl were found as a singlet at 2325 cm−1 and a doublet at 2422 and 2446 cm−1. The presence of a strong signal in the bending region and a singlet and a doublet in the stretching region are in agreement with a tridentate coordination of BH4− units to the La-metal,41 and consistent with the coordination of BH4− units to RE-metal in the reported structure of LiCe(BH4)3Cl.29 These bands are similar for the samples with RE = Ce, Pr, Nd. Additionally in the Nd sample there is a signal at 530 cm−1, which can arise from the NdOCl in the starting material.42 The IR spectra of the Sm, Gd, Tb and Er samples are similar, with a few additional bands in the B–H stretching and bending regions, which can be assigned to the respective RE(BH4)3 compound. From the measurements it is apparent that the strongest stretching modes of the RE(BH4)3 are situated in the same region as for LiBH4, but an additional weak band is seen in the region 2500–2560 cm−1. The bending modes for RE(BH4)3 are shifted from LiBH4, and thereby more easily distinguished. They are found around 1125 and 1190 cm−1. The band at 1190 cm−1 seems to be an unresolved doublet. The bands shift slightly in the samples of different lanthanides.
In the Eu sample the major signals are assigned to LiBH4, and there is only one broad peak in the B–H bending region (1160 cm−1) which could be assigned to a Eu–B–H compound. Both the Yb and Lu sample contain peaks similar to LiSc(BH4)4, confirming the relationship seen by PXD. The bending modes for the Lu sample are observed as an intense band at 1210 cm−1, together with two weaker bands at 1110 and 1330 cm−1. The stretching region is composed of several peaks, but the most apparent peaks are the doublets at 2470, 2250 and 2190 cm−1, respectively.
 | ||
| Fig. 2 IR data for RE = La, Eu, Er, Lu and LiBH4. | ||
Thermal properties
RE = La, Ce, Pr and Nd. The thermal decomposition of these four composites has been followed by TG/DSC measurements, and found to be similar (Fig. 3). The observed events are divided into three temperature regions. The first temperature region, 50–150 °C, includes a single broad endothermic signal in each sample, without any mass loss. The second temperature region, 150–230 °C, includes two partly overlapping but distinguishable endothermic events, one smaller broad event, and one larger sharp event. These reactions are accompanied by a mass loss of about 1.0–1.4 wt%. The third temperature region, 230–350 °C, includes two sharp endothermic peaks followed by a broad featureless endothermic event. These endothermic events are accompanied by a two-step mass loss in the range 4.0–4.3 wt%. Above 350 °C no additional events are observed up to 400 °C which is the maximum temperature of the experiment. The Nd sample had a different behavior in the third temperature region, which could be explained by the presence of NdOCl. Rest gas analysis indicates only hydrogen in the desorbed gas with no traces of diborane.
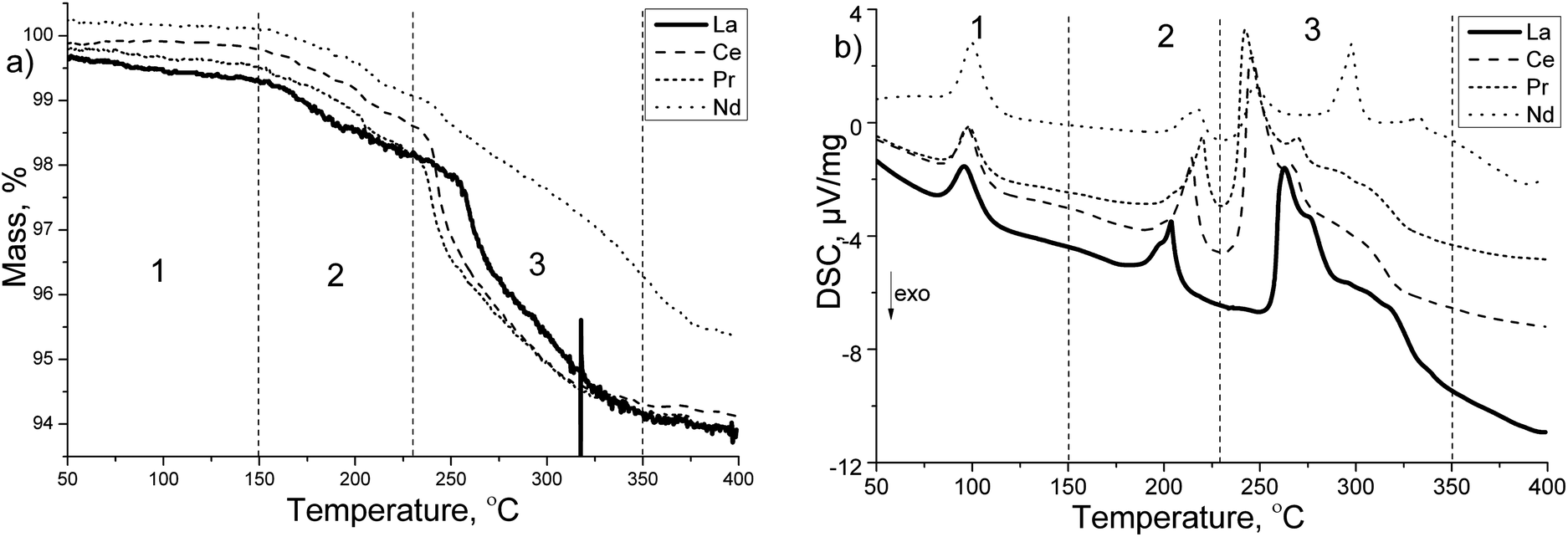 | ||
| Fig. 3 TG/DSC measurement performed on the RECl3 + 6 LiBH4 samples, with RE = La (solid bold), Ce (dashed), Pr (short dashed), Nd (dotted). (a) The TG signal given in mass% and (b) the DSC signal given in μV mg−1. The events are divided into three temperature regions: (1) from 50 to 150 °C, (2) from 150 to 230 °C and (3) from 230 to 350 °C. | ||
The endothermic event in region 1 is ascribed to the orthorhombic-to-hexagonal phase transition of LiBH4. The signal's broadening and the decrease in temperature compared to pure LiBH4 (115 °C), could be ascribed to a Cl-substitution in LiBH4.43
Comparing the behavior in region 2 to the reported thermal behavior of LiCe(BH4)3Cl by Frommen et al.,29 the broad signal and the mass loss can be ascribed to partial decomposition of LiRE(BH4)3Cl, while the sharp signal seems to be characteristic for the composite mixture of LiRE(BH4)3Cl and LiBH4, since it is not observed for the pure compounds. Gennari et al.34 also observed this large endothermic signal at 220 °C for their composite mixture of CeCl3 + 6 LiBH4, however they do not give any explanation to its presence. The temperature for the events in this region (2) are gradually increasing going from La to Nd. In the third region, the first endothermic reaction can be ascribed to the decomposition of the LiRE(BH4)3Cl phase. The second endothermic event could be ascribed to the melting of LiBH4, and the featureless endothermic event to the decomposition of LiBH4.
The decomposition of LiRE(BH4)3Cl and LiBH4 has been proposed to follow reaction (1)29 and (2),7 respectively:
 | (1) |
 | (2) |
According to literature, pure LiBH4 only desorbs 11 % (i.e. 2.0 wt% H) of its available hydrogen content up to 400 °C,34 which corresponds to about 0.35 wt% H in the 1 : 6 composites, and according to reaction (1) the mass losses from decomposition of LiRE(BH4)3Cl are 2.90–2.95 wt% for RE = La–Nd in the respective composites. Thus the observed mass losses of up to 5.6 wt% (in the Ce-sample) suggests that all borohydride species are decomposed, which indeed confirms that the presence of the RE-compound destabilizes the excess LiBH4. However, according to reactions (1) and (2), a theoretical mass loss of 5.35 wt% can be achieved in the 1:6 composite mixtures. Since the observed mass losses exceed this value, the total reaction could be a combination of these two reactions, in which REH2 reacts with the boron from LiBH4 forming additional REB6:
 | (3) |
A mass loss of 5.6 wt% can be achieved by reaction (3). A sample of LaCl3 + 6 LiBH4 annealed at 400 °C for 20 h showed LaB6 as the only La-containing crystalline compound, along with LiCl and LiH, with weight fractions corresponding to reaction (3), thus supporting this reaction.
RE = Gd, Tb, Er and Lu. The thermal decomposition of the Gd, Tb, Er and Lu samples resembles the behavior of the La, Ce and Pr systems. The DSC curves in Fig. 4a show one broad and one sharp endothermic peak in the temperature region 160–220 °C. There is a small mass loss of about 1 % associated with those events. The broad endothermic event is attributed to amorphization and partial decomposition of the parent borohydride phases whereas the sharp endothermic peak is characteristic for the composite mixtures containing excess LiBH4. Note that this sharp endothermic peak is absent in samples obtained from stoichiometric mixtures of RECl3 and LiBH4 in a molar ratio of 1 : 3, i.e. without excess of LiBH4.25,26,29 The onset temperatures for the partial decomposition or amorphization of the parent borohydride phases are shifted to lower temperatures with increasing atomic number. This trend is different from the earlier lanthanide borohydrides La, Ce and Pr, for which temperatures associated with their partial decomposition were found to increase with increasing atomic number.
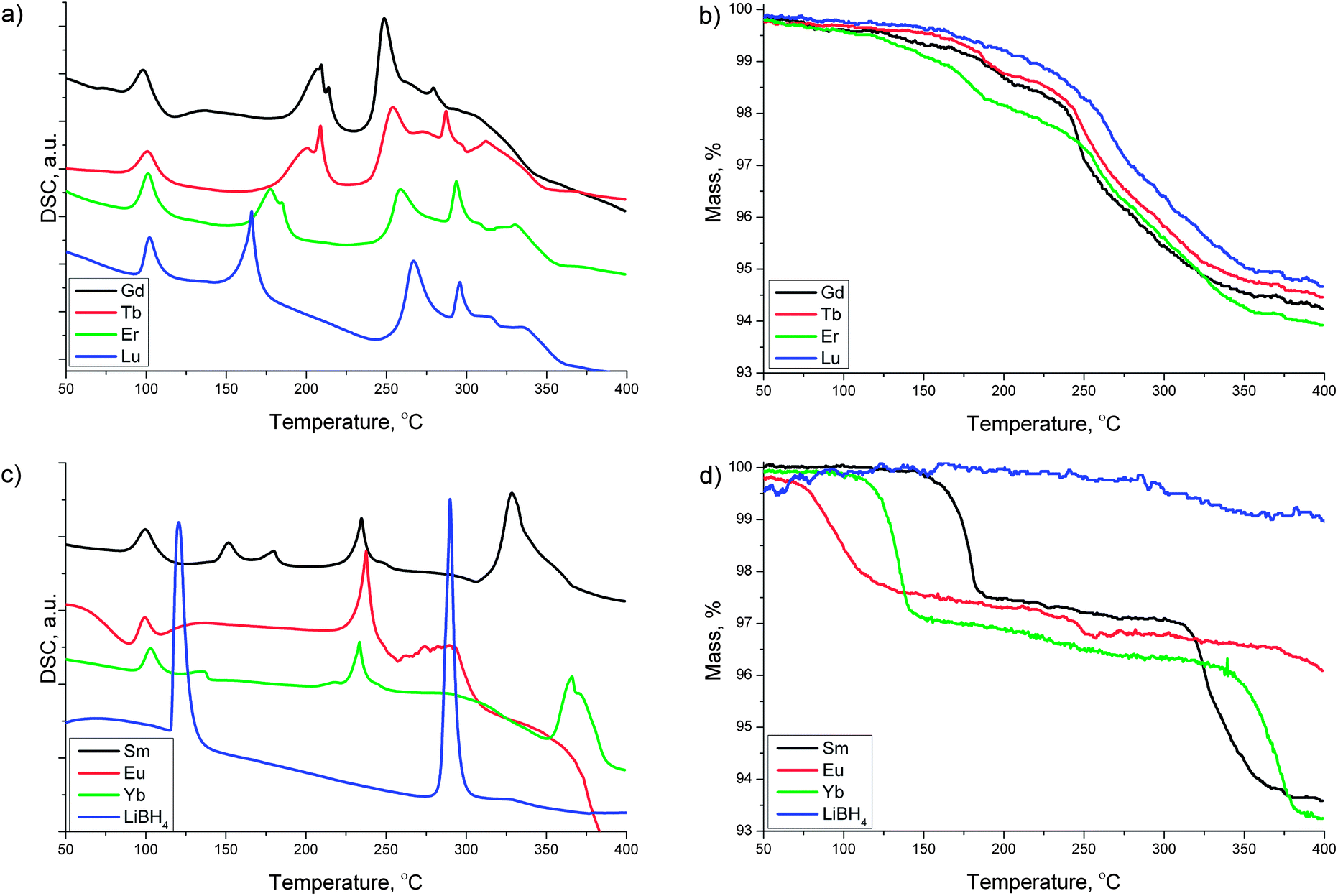 | ||
| Fig. 4 TG/DSC measurements for the RECl3 + 6 LiBH4 mixtures, with RE = Sm, Eu, Gd, Tb, Er, Yb and Lu. (a) DSC trace for the Gd, Tb, Er and Lu samples, (b) TG signal for the Gd, Tb, Er and Lu samples, (c) DSC trace for the Sm, Eu and Yb samples, (d) TG signal for the Sm, Eu and Yb samples. TG/DSC measurement for LiBH4 are added to (c) and (d) for comparison. | ||
Between 250 and 350 °C the DSC traces for Gd, Tb, Er and Lu (Fig. 4a) show two sharp endothermic peaks followed by a broad endothermic event coinciding with the main two-step mass loss (Fig. 4b). The first sharp endothermic event around 250–270 °C could be ascribed to the major decomposition of the borohydride phase whereas the sharp endothermic signal around 275–290 °C could have its origin in the melting of LiBH4. Finally, the broad featureless event around 320–350 °C could be caused by the decomposition of the remaining LiBH4 in the composite mixture. The DSC signals in the region between 250 and 350 °C are shifted to higher temperatures going from Gd to Lu. This behavior is again opposite to what is seen for the early lanthanides La, Ce and Pr.
RE = Sm, Eu and Yb. The composite mixtures containing Sm, Eu and Yb behave quite differently from the other RE samples, and their TG/DSC traces have been summarized in Fig. 4c and d, respectively. Our previous work on the Yb–BH4 system has shown that this element has the ability to form compounds in different oxidation states,23 thus the thermal properties are a combination of reduction/thermal decomposition reactions which results in a complex behavior. In the present RECl3–6LiBH4 mixtures containing Sm, Eu and Yb, one major mass loss is observed at temperatures below 200 °C. For Yb we have found from in situ SR-PXD data that the Yb(BH4)3 formed after ball-milling is reduced to Yb(BH4)2 upon heating around 100 °C.23 This reduction is accompanied by a release of diborane gas. We assume that the first major mass loss observed in the TG traces below 180 °C, 150 °C and 100 °C for the Sm, Yb and Eu samples, respectively, is associated with the same reduction from the trivalent to the divalent state of the RE metal and release of diborane gas, as these RE elements have a tendency to take a divalent state (see Fig. 4d). For Eu this mass loss is accompanied by a broad exothermic DSC event around 100 °C, while in the Yb sample the mass loss is associated with an endothermic DSC peak at 135 °C (see Fig. 4c).
Going to higher temperatures, we observe two DSC events in the 150–180 °C region for the Sm-sample: one endothermic signal at 150 °C with no mass loss, and a second endothermic peak at 180 °C corresponding to the first major mass loss. Around 230 °C the Sm, Eu and Yb-samples exhibit similar endothermic DSC peaks that are not associated with any significant mass loss. These events could be attributed to an amorphization of either the divalent RE(BH4)2 borohydride or LiBH4. This assumption is supported by the disappearance of diffraction peaks belonging to RE(BH4)2 and LiBH4 in the in situ SR-PXD data in the same temperature region (see below).
Increasing the temperature further, there is an endothermic feature in the Sm sample in the temperature region of 300–400 °C which is similar to the features mentioned above for the other samples in the temperature region of 250–350 °C, including a two-step mass loss. These features can also be observed in the Yb sample above 350 °C, while they are not observed in the Eu sample up to 400 °C.
RE = La. Due to experimental conditions the maximum temperature of the experiment was about 300 °C. The SR-PXD data collected during heating under dynamic vacuum showed that the ball milled sample contained larger amounts of the starting material LaCl3, as opposed to in the annealed samples presented above. The complete experimental data set is presented in Fig. 5, in which the three temperature regions used in the interpretation of the TG/DSC data are shown. Fig. 6 consists of four plots (a–d) showing selected diffraction patterns to illustrate the changes in the respective temperature region. Fig. 7 shows selected peak positions and their integrated intensities as a function of temperature.
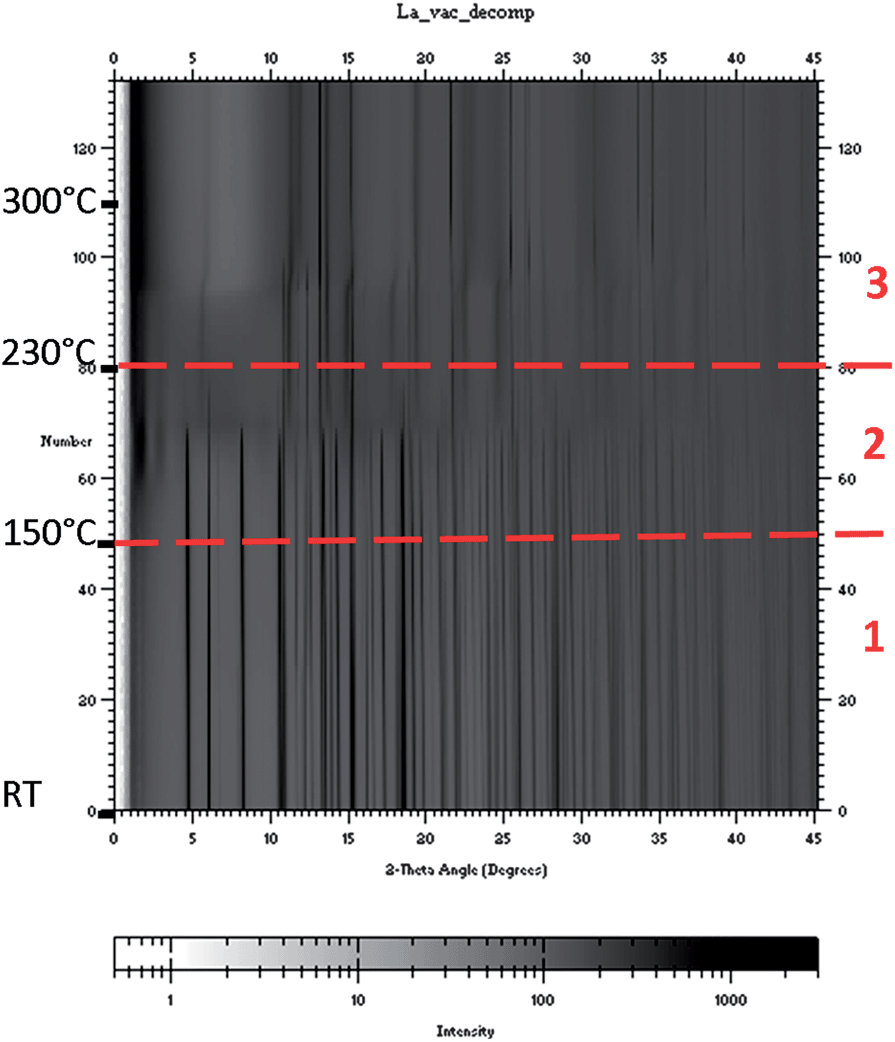 | ||
| Fig. 5 In situ SR-PXD patterns during heating of the LaCl3 + 6 LiBH4 sample, λ = 0.69411 Å. Each consecutive pattern is given on the y-axis, 2θ on the x-axis, and the intensity is given by the color map at the bottom. The temperature regions defined by TG/DSC measurements are given by horizontal dashed red lines, with the corresponding region number on the right-hand side. The maximum temperature of the experiment was around 300 °C, and temperatures are inserted on the left-hand side. | ||
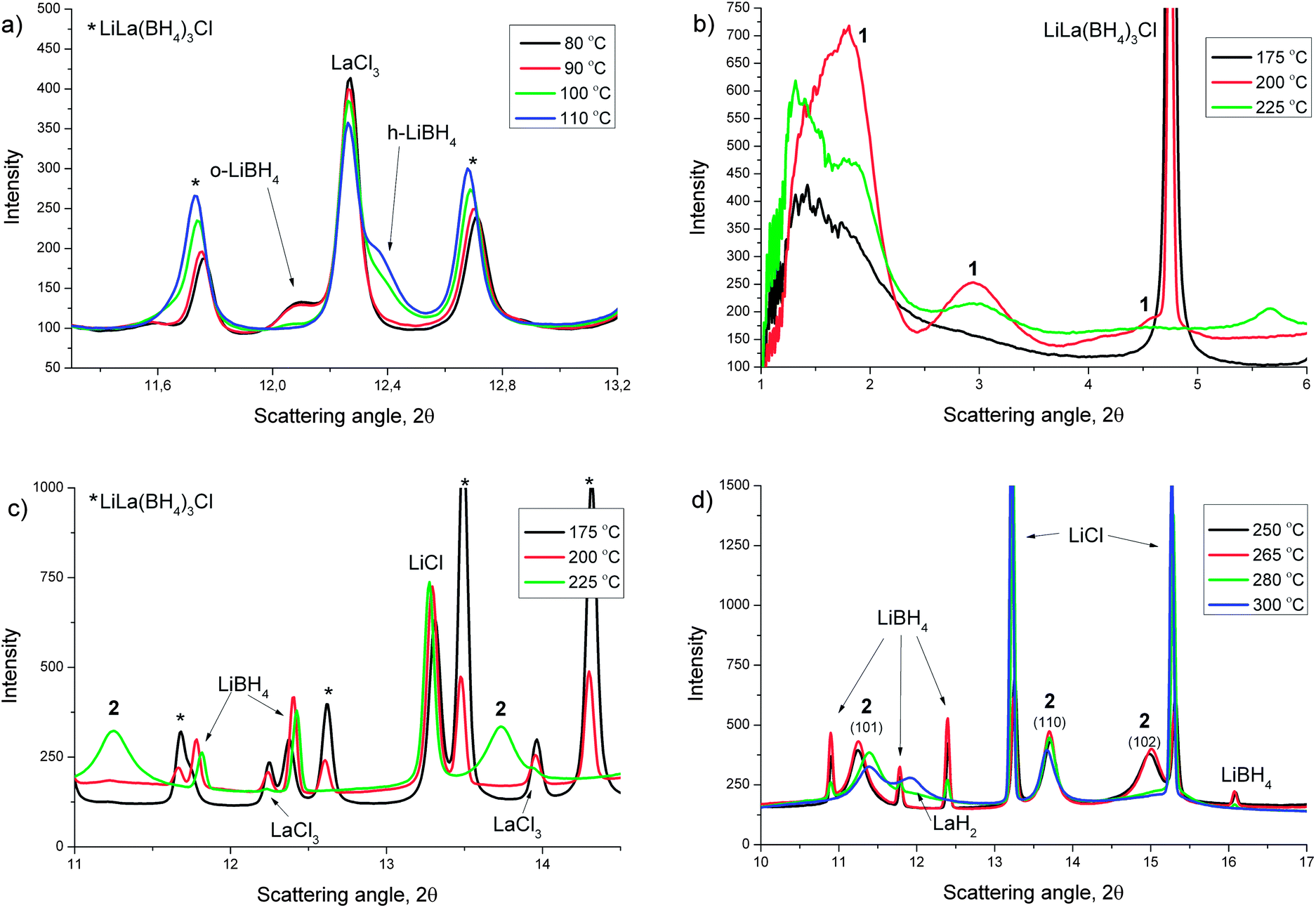 | ||
| Fig. 6 In situ SR-PXD patterns at selected temperatures during heating of a LaCl3 + 6 LiBH4 mixture. (a) Phase transition of LiBH4 in temperature region 1, (b) low angle data in temperature region 2, “1” represents peaks from intermediate 1, (c) higher angle data in temperature region 2, “2” represents the peaks from intermediate 2 and (d) temperature region 3, “2” represents intermediate 2. λ = 0.69411 Å. | ||
 | ||
| Fig. 7 Selected peak positions and their area from the in situ SR-PXD patterns during heating of LaCl3 + 6 LiBH4. The temperature regions defined by DSC are marked by bold numbers and vertical dashed lines, 1 ≤ 150 °C, 2 = 150–230 °C and 3 ≥ 230 °C. (a) Peak positions for LiBH4 and LiCl, (b) peak positions for the La-containing phases, (c) peak area of the LiBH4 phases and LiCl, (d) peak area for the La-containing phases. Where error bars are not visible the uncertainty is within the symbol. | ||
The data show the orthorhombic-to-hexagonal phase transition of LiBH4 around 100 °C (Fig. 6a). After the phase transition the Bragg peak positions of h-LiBH4 exhibit a shift to higher angles with increasing temperature (Fig. 7a) indicating chloride substitution.43 The small decrease in peak intensity of LiCl (Fig. 7c, 85–105 °C) complies with this. Additionally, the relative peak intensities show a faster conversion of LaCl3 to LiLa(BH4)3Cl after the phase transition of LiBH4 (Fig. 7d). The diffraction data in temperature region 2 (150–230 °C) show that the intensity of the Bragg reflections for LiLa(BH4)3Cl start to decrease (Fig. 7d), along with the appearance of some broad peaks at low angle (Fig. 6b), indicating a phase with a large unit cell, named intermediate 1, which has not been successfully identified yet. Simultaneously, an increased chloride substitution was observed in LiBH4 (Fig. 7a, 180–200 °C). These events were followed by the evolution of a new set of broad diffraction peaks (Fig. 6c, 2), which were indexed with a primitive tetragonal structure with a = 4.11 Å and c = 6.97 Å. This phase has been named intermediate 2 and has not yet been further characterized. When the peaks for 2 evolve, the diffracted intensity from 1 decrease (Fig. 7d).
The SR-PXD data corresponding to temperature region 3 (230–300 °C) (Fig. 6d) show that the diffraction peaks from intermediate 2 exhibit an anisotropic behavior (around 270 °C), which can be assigned to a contraction along the tetragonal axis on heating. Firstly the peaks hk0 are shifted marginally to higher angles (Fig. 6d, 2 (110)), secondly the peaks hk1 (Fig. 6d, 2 (101)) have moved to higher angles and broadened, and finally the peaks hk2 (Fig. 6d, 2 (102)) exhibit a larger shift to higher angles and broaden. Simultaneously the intensity of the Bragg peaks from LiBH4 start to decrease and the peaks from LiCl rapidly increase (Fig. 7c). After this, 2 started to decompose and LaH2 was formed (Fig. 7d). The decomposition of 2 was not completed at the maximum temperature of the experiment.
The in situ SR-PXD show that there are more reactions occurring in the composite than what is evident from the DSC, and another observation was that crystalline LiBH4 actually disappeared at a lower temperature than the decomposition of 2 to LaH2.
The in situ SR-PXD experiment of a LaCl3 + 6 LiBH4 sample under 0.5 MPa H2 backpressure show the same features as under dynamic vacuum up to temperature region 3 (Fig. S2 and S3 in ESI†). However, the major differences compared to the vacuum experiment are that the diffraction peaks for LaH2 did not appear, and that the peaks from 2 exhibit a different behavior. The intensity of the l = 2n peaks decreased, while the intensity of all other peaks have increased. The peak position of all peaks with l ≠ 0 are shifted to higher angles, while peaks with l = 0 keep their position. It appears that this phase is stabilized by hydrogen backpressure.
RE = Er. The in situ SR-PXD data from thermal decomposition of the Er sample in dynamic vacuum are given in Fig. 8. At RT the diffraction pattern for the Er sample includes peaks from β-Er(BH4)3 and LiCl as major phases, in addition to small peaks from o-LiBH4 and α-Er(BH4)3. At 105 °C the phase transition from o-LiBH4 to h-LiBH4 was observed. In the temperature region 170–210 °C Bragg peaks from Er(BH4)3 decreased and disappeared, while the background intensity increased with a modulation indicating amorphous compounds. Additionally the intensity of LiCl decreased, while peaks from h-LiBH4 shifted to higher angles and sharpened, indicating a Cl-substitution in LiBH4.43 In the temperature region 240–270 °C a set of broad peaks appeared. These peaks could originate from an intermediate decomposition product, similar to what we have observed in the La-system although the peak positions and intensities are different from those seen in the La-system. Simultaneously the LiCl peaks sharpened and decreased and LiBH4 increased in intensity. Increasing the temperature further (290–370 °C) resulted in increased peak intensity of LiCl, the broad peaks disappeared, peaks belonging to ErH2 appeared, and the LiBH4 peaks disappeared suddenly at 330 °C. This confirms that the broad peaks can be assigned to an intermediate Er-containing compound, as it transforms into ErH2. Only a slight sharpening of ErH2 peaks was observed up to the maximum temperature of 400 °C.
 | ||
| Fig. 8 (a) Contour plot of the in situ SR-PXD measurement on the ErCl3 + 6 LiBH4 mixture with dynamic vacuum. (b) Diffraction patterns at selected temperatures. λ = 0.694118 Å. | ||
RE = Gd. The Gd sample (Fig. S5†) give Bragg peaks from the starting material GdCl3 after ball milling, thus the metathesis reaction was not complete. However during heating the reaction is completed below 150 °C seen by disappearance of Bragg peaks from GdCl3 along with increased intensity in the reflections from both Gd(BH4)3 and LiCl. When Gd(BH4)3 disappeared from the diffraction pattern, peaks belonging to LiGd(BH4)3Cl appeared in a small temperature region of 10 °C around 230 °C. Apart from this the decomposition follows the same path as for the Er sample.
RE = Sm. The in situ SR-PXD decomposition data (Fig. 9) for the Sm sample are different from the Er- and Gd-samples, and thus in agreement with what is expected from the TG/DSC measurements. The as-milled sample included a multitude of crystalline phases. The major phases were α/β-Sm(BH4)3 (39.2(2) wt%), SmCl3 (15.6(2) wt%) and LiCl (34.9(2) wt%), while LiSm(BH4)3Cl (1.9(2) wt%), LiBH4 (1.7(5) wt%) and SmOCl (6.8(2) wt%) were present as minor phases. Up to 120 °C the phase transition of LiBH4 occurred in parallel with a decrease in intensity of SmCl3 and increasing intensity of Sm(BH4)3. A transition from Sm(BH4)3 to LiSm(BH4)3Cl is observed around 150 °C, consistent with the endothermic event without mass loss in the TG/DSC measurement. Above 100 °C a new set of peaks started to evolve, which increased in intensity while the peak intensity of LiSm(BH4)3Cl decreased and disappeared at around 180 °C. The set of new peaks could be indexed on an orthorhombic unit cell a = 6.99, b = 8.45 and c = 7.59 Å at approximately 200 °C. Systematic absences proposed that the compound crystallizes in the space group Pbcn (no. 60). A structure model was obtained with FOX in this space group, where the unit cell contains 4 Sm atoms and 8 BH4-tetrahedra units. During simulations Li and Cl atoms were included, but they merged with the Sm atoms, thus indicating that this phase has the composition Sm(BH4)2. Synthesis of a deuterided sample for powder neutron diffraction to locate the H(D) atoms, was not attempted due to the extremely large neutron absorption cross section of natural Sm (5900 barn).
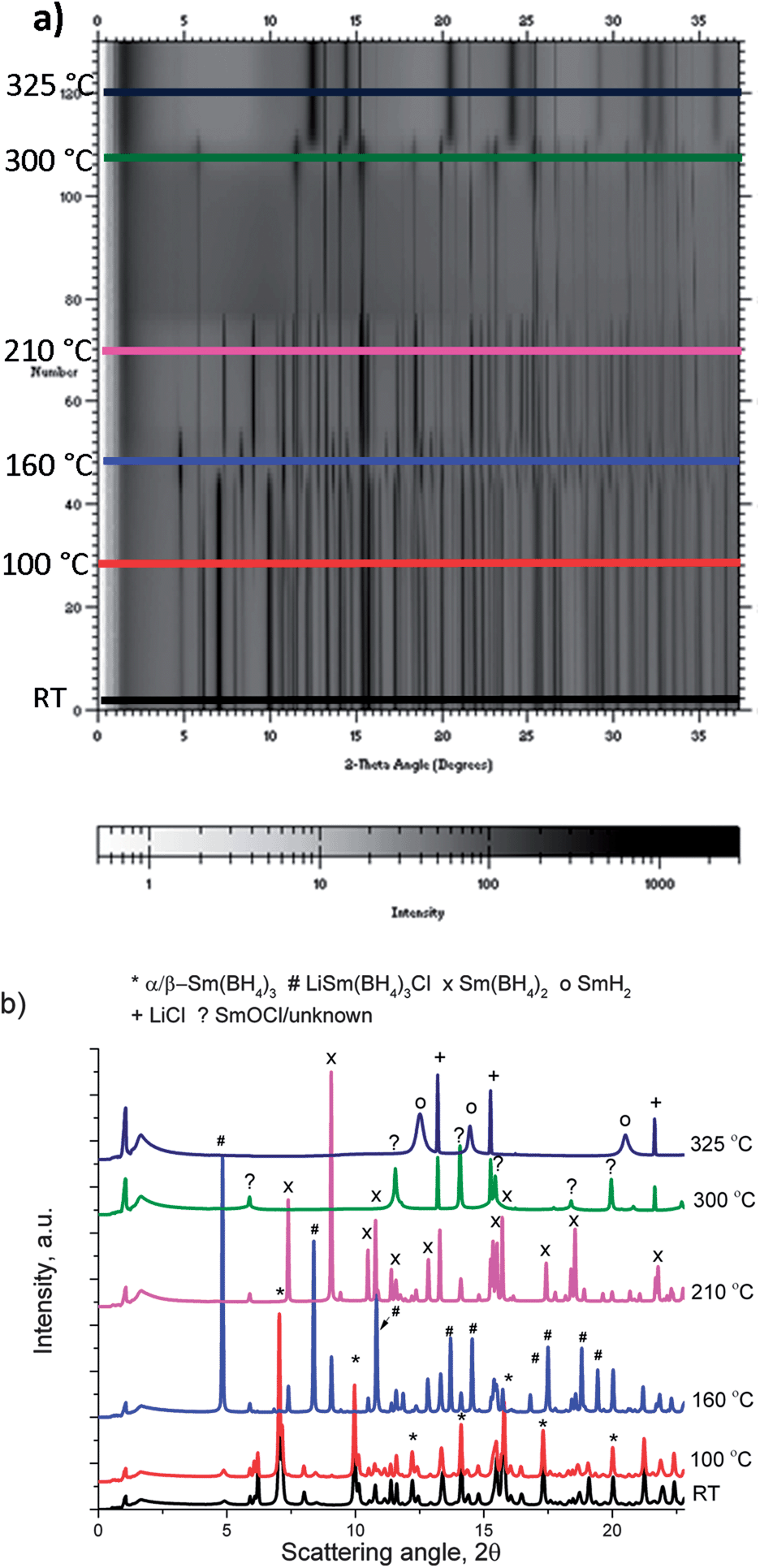 | ||
| Fig. 9 (a) Contour plot of the in situ SR-PXD measurement on the SmCl3 + 6 LiBH4 mixture with dynamic vacuum. (b) Diffraction patterns at selected temperatures. λ = 0.694118 Å. | ||
The Rietveld fit of Sm(BH4)2 is presented in Fig. 10. Four phases were included and the refined phase composition was 41.4(1) wt% Sm(BH4)2, 5.9(1) wt% SmOCl, 19.5(2) wt% Li(BH4)0.62Cl0.38 and 33.2(1) wt% LiCl. The amount of Sm (39.2 wt%) is in good agreement with the nominal amount of Sm (40.4 wt% disregarding initial oxygen content). The reliability factors in the final refinement were: Rwp = 3.18% and Rp = 2.69%. The refined unit cell dimensions at 200 °C are a = 6.9848(1) Å, b = 8.4464(1) Å and c = 7.5891(1) Å. The structure of Sm(BH4)2 can be described by Sm(BH4)6 octahedra sharing edges with two other octahedra in an angle of 111.8°, and thus building chains in the c-direction (Fig. 11). Each chain of octahedra is connected via corner sharing to four others in the (110)-planes. The octahedral environment of BH4-units around Sm is distorted with trans angles ranging from 167.8 to 173.0° and Sm–B distances of 2.996, 3.011 and 3.061 Å. Each BH4-unit is surrounded by 3 Sm atoms in a distorted trigonal planar environment, where the Sm–B–Sm angle in the edge sharing octahedra is 98.3°. This structure-type is not reported earlier for metal borohydrides. Tables with crystallographic data and interatomic distances and angles can be found in the ESI (Tables S7–S8†).
 | ||
| Fig. 10 The Rietveld refinement of in situ SR-PXD data at around 200 °C for the SmCl3 + 6 LiBH4 sample. Experimental data as circles, calculated diffraction pattern as a red line, tick marks for LiCl (top), LiBH4, SmOCl and Sm(BH4)2 (bottom), respectively, and the difference plot. The wavelength was: 0.694118 Å. | ||
 | ||
| Fig. 11 Representative views of the structure of Sm(BH4)2, (a) viewed along the c-axis, (b) 3 Sm-(BH4)6 octahedral units along the chain in c. Sm atoms as large grey balls, BH4 tetrahedra represented with teal faces and Sm-(BH4)6 octahedra represented with red faces. | ||
This confirms the suspicion from the TG/DSC measurements that Sm is reduced from a trivalent to a divalent state during thermal decomposition. At around 230 °C, Sm(BH4)2 disappeared from the diffraction pattern, and only crystalline SmOCl, LiBH4 and LiCl were observed. The background intensity increased over the whole pattern, indicating a melting. Shortly thereafter peaks from LiBH4 disappeared. Then a set of broad peaks appeared in the same positions as the sharp SmOCl reflexes, along with decreasing background intensity. Above 300 °C these broad peaks and the signal from SmOCl disappeared while broad peaks from SmH2 evolved in the diffraction patterns.
RE = Yb. The measurement on the Yb-sample (Fig. S7†) showed the same path as we have reported earlier.23 The trivalent Yb-borohydride phases (LiYb(BH4)4−xClx as major phase, α/β-Yb(BH4)3 as minor phases) decomposed to the divalent tet-Yb(BH4)2−xClx phase around 130 °C. However this phase disappeared completely around 230 °C in the present sample with excess LiBH4, compared to the peritectic behavior observed previously for the 3 LiBH4 + YbCl3 sample.
Thermal decomposition against H2 backpressure have been shown to alter the decomposition pathway of borohydrides,34 therefore in situ SR-PXD during decomposition with 0.5 M Pa H2 backpressure where conducted for the samples with Er, Gd, Sm and Yb. In these experiments the reactions occurred at higher temperatures than for the dynamic vacuum experiments; but there were no other major changes. The only observed difference is the absence of the crystallization of ErH2 and SmH2 in the Er- and Sm-sample, respectively.
RE = La. Within a few minutes the intensity of the Bragg peaks belonging to LaH2 increased in the sample decomposed in vacuum (Fig. S2†), while only minor peaks from LaH2 appeared for the sample decomposed under 0.5 MPa hydrogen backpressure (Fig. S3†). During cooling under 10 MPa H2 pressure, Bragg peaks for h-LiBH4 reappeared in the diffraction pattern at the same temperature as they disappeared during heating. The peaks belonging to LiCl shifted to higher angles due to thermal contraction, while the peak positions of LaH2 stayed at the same angle down to RT, indicating some hydrogen absorption compensating the thermal contraction.
In addition to in situ SR-PXD studies, the thermal decomposition and rehydrogenation was also followed ex situ. For this purpose, a ball milled sample of LaCl3 + 6 LiBH4 was heated to 430 °C under static vacuum and subsequently rehydrogenated at 300 °C under 10 MPa H2. The total mass loss of the milled sample measured by TG/DSC was 5.3 wt%, and the TG/DSC of the decomposed sample showed zero mass loss, confirming full decomposition. TG/DSC of the rehydrogenated sample showed a mass loss of 0.95 wt% between 240 and 300 °C, corresponding to 18% of the full H-capacity. LiBH4 was not observed in the IR spectra of the rehydrogenated sample, however there are features in both the B–H bending and stretching region. This could indicate the presence of some B–H containing compound. The sample crystallinity was too poor to identify the phase by PXD. A different LaCl3 + 6 LiBH4 sample decomposed under the same conditions and rehydrogenated at 415 °C under 10 MPa H2 exhibited a much higher mass loss of 1.9 wt% (36% of full H-capacity) in TG/DSC between 275 and 350 °C. The DSC trace showed an endothermic event around 100 °C and another at 275 °C followed by a broad featureless endothermic signal in the temperature range 280–350 °C which could be assigned to the phase transition of LiBH4, melting of LiBH4 and decomposition of LiBH4, respectively. PXD revealed very poor crystallinity in the sample, but IR spectra from the rehydrogenated sample also indicated the presence of LiBH4 or another B–H containing phase. The TG/DSC measurements and IR spectra of these rehydrogenated samples can be found in the ESI, Fig. S8 and S9,† respectively.
RE = Er. A sample of ErCl3 + 6 LiBH4 was decomposed ex situ at 430 °C for 2 hours to assure complete decomposition, and then rehydrogenated at 415 °C and 10 MPa H2 for 20 hours. The rehydrogenated sample showed a mass loss of about 1.4 wt% in the temperature region 300–400 °C (determined by TG/DSC), and the DSC trace showed events which could be assigned to the presence of LiBH4 (Fig. S10†). The observed mass loss corresponds to a 25% rehydrogenation. An IR spectrum (Fig. S11†) of the rehydrogenated sample exhibits weak bands similar to those of LiBH4. Despite the fact that LiBH4 is destabilized, the rehydrogenation conditions are reduced from above 600 °C and 15.5 MPa (ref. 7) to 415 °C and 10 MPa. This indicates that the kinetics play an important role in the decomposition/rehydrogenation reaction of LiBH4.
Discussion
RE = La, Ce, Pr and Nd
The thermal decomposition of the composites with LiRE(BH4)3Cl and LiBH4 are complex. When mixed with LiBH4, LiLa(BH4)3Cl starts to decompose below 200 °C, and the in situ SR-PXD data shows the formation of at least two different unknown intermediate phases. The second intermediate phase decomposes at 260 °C, similar to the decomposition reported for the pure LiRE(BH4)3Cl compounds.29–31 The remaining LiBH4 in the composite decomposes and release all hydrogen below 350 °C, which is at much lower temperature than pure LiBH4 (onset at 380 °C, max desorption rate at 430 °C).7The composites drastically reduce the temperature needed to rehydrogenate LiBH4. Mauron et al.7 has shown that the decomposed sample of pure LiBH4 could be rehydrogenated by applying 15.5 MPa H2 pressure at 600 °C, while the composite with La-borohydride show a partial (18%) rehydrogenation at 10 MPa and 300 °C which increased by a factor of two (to 36%) by raising the temperature to 415 °C in this work.
Two unknown phases (intermediate 1 and 2) are observed during the in situ SR-PXD measurement of the 6 LiBH4–LaCl3 composite. Intermediate 1 was seen as broad features at low angles indicating a large unit cell, however indexing was not successful. Intermediate 2 had more numerous and more intense peaks. The peaks could be indexed by a tetragonal unit cell with lattice constants a = 4.11 Å and c = 6.97 Å. The only known La-compound which is reported with similar unit cell is LaOCl (P4/nmm, a = 4.11, c = 6.88 Å).44 The starting material has been checked by IR, and there is no evidence of LaOCl or other oxide vibrations. The phase also appeared during thermal decomposition against 0.5 MPa hydrogen backpressure. This rules out any significant oxygen leakage into the system. The fact that this phase is more stable during thermal decomposition against 0.5 MPa hydrogen backpressure indicates that it contains hydrogen. Furthermore the relative peak intensities of the peaks do not match that of the reported LaOCl structure. Finally LaOCl should be thermally stable up to 500 °C (ref. 45) and from Fig. 7d the decomposition of intermediate 2 starts at 280 °C. Thus it can be argued that intermediate 2 is not LaOCl. The correlation between the diffracted intensities of LiLa(BH4)3Cl, 1, 2 and LaH2 (Fig. 7d) clearly points to the fact that the two unidentified phases are intermediate decomposition products of LiLa(BH4)3Cl.
Frommen et al.29 reported at least three steps in the decomposition of LiCe(BH4)3Cl by DSC when the heating rate was increased and the events were more separated. The composite formation with LiBH4 could change the thermal stabilities of these intermediate phases, which both results in a separation of the events in our TG/DSC measurement and the observation of these phases in the in situ SR-PXD data. Gennari et al.31 have studied the ball milled composite of CeCl3 + 3 LiBH4, and reported several peaks of an unknown phase, similar to intermediate 2, in a sample obtained after isothermal decomposition at 200 °C, as well as after heating to 350 °C. Zhang et al.32 also observed similar diffraction peaks in similar ball milled CeCl3 + 3 LiBH4 composite after dehydrogenation at 450 °C, and after subsequent rehydrogenation at 450 °C, 10 MPa H2 for 24 h. However, after the second dehydrogenation the phase was not present. Both Gennari et al. and Zhang et al. proposed this as an intermediate phase during the decomposition of “Ce(BH4)3” (which they believed was the composition of the LiCe(BH4)3Cl phase). Furthermore, Gennari et al. suggested the formation of Ce2(B12H12)3 similar to the intermediates observed during thermal hydrogen desorption of LiBH4, Mg(BH4)2 and LiSc(BH4)4.46 The volume of the tetragonal unit cell found in the present work (intermediate 2) cannot fit a large structure such as Ce2(B12H12)3, but 1 might be such a phase. Samples with better crystallinity have to be obtained in order to identify and solve the crystal structures of the two intermediate phases.
RE = Sm, Eu, Gd, Tb, Er, Yb and Lu
Even though the lanthanide borohydrides have different crystal structures, their decomposition behavior and their destabilizing effect on LiBH4 seems to be rather similar. In the 6 LiBH4 + RECl3 mixtures containing Gd, Tb, Er and Lu, the RE-borohydride decomposes around 250 °C releasing hydrogen, followed by a hydrogen release from the remaining LiBH4, below the decomposition temperature of pure LiBH4. An exception from this is found in the composites containing Sm, Eu and Yb. In these cases, diborane gas release was observed by mass spectroscopy below 200 °C, when the trivalent borohydride is reduced to a divalent borohydride, which finally decomposes at higher temperatures (above 300 °C). The destabilization of LiBH4 is observed only after this decomposition. This confirms that it is the formation of REH2 which destabilize LiBH4 as suggested by Gennari et al.34 Another interesting effect is that the trivalent RE-BH4 phases seem to become amorphous or melt before decomposition when additional LiBH4 is present in the system. Finally it seems that the additional LiBH4 stabilizes an unknown intermediate compound, which is presented with the broad peaks in the Er-sample above 240 °C.Conclusions
Mechanochemical milling of RECl3 + 6 LiBH4 results in new compounds of LiRE(BH4)3Cl (RE = La, Ce, Pr, Nd, Sm, Gd), RE(BH4)3 (RE = Sm, Gd, Tb, Er, Yb) and/or LiRE(BH4)4 (RE = Yb, Lu) with LiBH4 and LiCl. These composites destabilize the remaining LiBH4, desorbing most of the hydrogen below 400 °C. Partial rehydrogenation is observed with 1.8 wt% hydrogen desorption for the rehydrogenated sample in the La-system. IR spectroscopy indicates that the rehydrogenated sample contains LiBH4 or another BH4-containing phase. Despite the fact that LiBH4 is destabilized by composite formation with a RECl3 the rehydrogenation is possible at milder conditions, which indicates that the kinetics play an important role in the decomposition/rehydrogenation reaction of LiBH4.Acknowledgements
The Research Council of Norway is acknowledged for financial support through the FRIENERGI project and the SYNKNØYT program. We acknowledge the skilful assistance from the staff of SNBL at ESRF, Grenoble in France. The work was supported by the Danish National Research Foundation, Center for Materials Crystallography (DNRF93), the Danish Strategic Research Council (the research project HyFillFast).References
- A. Züttel, A. Borgschulte and S. I. Orimo, Scr. Mater., 2007, 56, 823–828 CrossRef PubMed.
- H. W. Li, Y. G. Yan, S. Orimo, A. Zuttel and C. M. Jensen, Energies, 2011, 4, 185–214 CrossRef CAS.
- L. H. Rude, T. K. Nielsen, D. B. Ravnsbæk, U. Bosenberg, M. B. Ley, B. Richter, L. M. Arnbjerg, M. Dornheim, Y. Filinchuk, F. Besenbacher and T. R. Jensen, Phys. Status Solidi A, 2011, 208, 1754–1773 CrossRef CAS.
- C. Li, P. Peng, D. W. Zhou and L. Wan, Int. J. Hydrogen Energy, 2011, 36, 14512–14526 CrossRef CAS PubMed.
- D. B. Ravnsbæk, L. H. Sørensen, Y. Filinchuk, F. Besenbacher and T. R. Jensen, Angew. Chem., Int. Ed., 2012, 51, 3582–3586 CrossRef PubMed.
- H. I. Schlesinger and H. C. Brown, J. Am. Chem. Soc., 1940, 62, 3429–3435 CrossRef CAS.
- P. Mauron, F. Buchter, O. Friedrichs, A. Remhof, M. Bielmann, C. N. Zwicky and A. Züttel, J. Phys. Chem. B, 2008, 112, 906–910 CrossRef CAS PubMed.
- P. Martelli, R. Caputo, A. Remhof, P. Mauron, A. Borgschulte and A. Züttel, J. Phys. Chem. C, 2010, 114, 7173–7177 CAS.
- K. Miwa, M. Aoki, T. Noritake, N. Ohba, Y. Nakamori, S. Towata, A. Zuttel and S. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 155122 CrossRef.
- M. D. Riktor, M. H. Sørby, K. Chlopek, M. Fichtner, F. Buchter, A. Züttel and B. C. Hauback, J. Mater. Chem., 2007, 17, 4939–4942 RSC.
- K. Chlopek, C. Frommen, A. Leon, O. Zabara and M. Fichtner, J. Mater. Chem., 2007, 17, 3496–3503 RSC.
- M. Chong, A. Karkamkar, T. Autrey, S. Orimo, S. Jalisatgi and C. M. Jensen, Chem. Commun., 2011, 47, 1330–1332 RSC.
- Y. Kim, S. J. Hwang, J. H. Shim, Y. S. Lee, H. N. Han and Y. W. Cho, J. Phys. Chem. C, 2012, 116, 4330–4334 CAS.
- G. Severa, E. Rönnebro and C. M. Jensen, Chem. Commun., 2010, 46, 421–423 RSC.
- D. Ravnsbæk, Y. Filinchuk, Y. Cerenius, H. J. Jakobsen, F. Besenbacher, J. Skibsted and T. R. Jensen, Angew. Chem., Int. Ed., 2009, 48, 6659–6663 CrossRef PubMed.
- R. A. Varin, L. Zbroniec, M. Polanski, Y. Filinchuk and R. Cerny, Int. J. Hydrogen Energy, 2012, 37, 16056–16069 CrossRef CAS PubMed.
- Y. Nakamori, K. Miwa, A. Ninomiya, H. W. Li, N. Ohba, S. I. Towata, A. Zuttel and S. I. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 045126 CrossRef.
- G. N. Schrauzer, Naturwissenschaften, 1955, 42, 438 CrossRef CAS.
- Y. Nakamori, H. W. Li, K. Kikuchi, M. Aoki, K. Miwa, S. Towata and S. Orimo, J. Alloys Compd., 2007, 446, 296–300 CrossRef PubMed.
- E. A. Nickels, M. O. Jones, W. I. F. David, S. R. Johnson, R. L. Lowton, M. Sommariva and P. P. Edwards, Angew. Chem., Int. Ed., 2008, 47, 2817–2819 CrossRef CAS PubMed.
- H. W. Li, S. Orimo, Y. Nakamori, K. Miwa, N. Ohba, S. Towata and A. Züttel, J. Alloys Compd., 2007, 446, 315–318 CrossRef PubMed.
- H. Hagemann, M. Longhini, J. W. Kaminski, T. A. Wesolowski, R. Cerny, N. Penin, M. H. Sørby, B. C. Hauback, G. Severa and C. M. Jensen, J. Phys. Chem. A, 2008, 112, 7551–7555 CrossRef CAS PubMed.
- J. E. Olsen, C. Frommen, M. H. Sørby and B. C. Hauback, RSC Adv., 2013, 3, 10764–10774 RSC.
- T. Sato, K. Miwa, Y. Nakamori, K. Ohoyama, H. W. Li, T. Noritake, M. Aoki, S. I. Towata and S. I. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 104114 CrossRef.
- C. Frommen, N. Aliouane, S. Deledda, J. E. Fonneløp, H. Grove, K. Lieutenant, I. Llamas-Jansa, S. Sartori, M. H. Sørby and B. C. Hauback, J. Alloys Compd., 2010, 496, 710–716 CrossRef CAS PubMed.
- D. B. Ravnsbæk, Y. Filinchuk, R. Cerny, M. B. Ley, D. Haase, H. J. Jakobsen, J. Skibsted and T. R. Jensen, Inorg. Chem., 2010, 49, 3801–3809 CrossRef PubMed.
- Y. S. Lee, J. H. Shim and Y. W. Cho, J. Phys. Chem. C, 2010, 114, 12833–12837 CAS.
- M. B. Ley, S. Boulineau, R. Janot, Y. Filinchuk and T. R. Jensen, J. Phys. Chem. C, 2012, 116, 21267–21276 CAS.
- C. Frommen, M. H. Sørby, P. Ravindran, P. Vajeeston, H. Fjellvåg and B. C. Hauback, J. Phys. Chem. C, 2011, 115, 23591–23602 CAS.
- M. B. Ley, D. B. Ravnsbæk, Y. Filinchuk, Y. S. Lee, R. Janot, Y. W. Cho, J. Skibsted and T. R. Jensen, Chem. Mater., 2012, 24, 1654–1663 CrossRef CAS.
- F. C. Gennari and M. R. Esquivel, J. Alloys Compd., 2009, 485, L47–L51 CrossRef CAS PubMed.
- B. J. Zhang, B. H. Liu and Z. P. Li, J. Alloys Compd., 2011, 509, 751–757 CrossRef CAS PubMed.
- E. M. Fedneva, V. I. Alpatova and V. I. Mikheeva, Zh. Neorg. Khim., 1964, 9, 1519–1520 CAS.
- F. C. Gennari, L. F. Albanesi, J. A. Puszkiel and P. A. Larochette, Int. J. Hydrogen Energy, 2011, 36, 563–570 CrossRef CAS PubMed.
- A. V. Skripov, A. Soloninin, M. B. Ley, T. R. Jensen and Y. Filinchuk, J. Phys. Chem. C, 2013, 117, 14965–14972 CAS.
- P. E. Werner, L. Eriksson and M. Westdahl, J. Appl. Crystallogr., 1985, 18, 367–370 CrossRef CAS.
- V. Favre-Nicolin and R. Cerny, J. Appl. Crystallogr., 2002, 35, 734–743 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, Los Alamos National Laboratory Report, 1994 Search PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- V. M. Goldschmidt, G. Lunde and T. F. W. Barth, Isomorphie und Polymorphie der Sesquioxyde: die Lanthanidenkontraktion und ihre Konsequenzen, Dybwad, Oslo, 1925 Search PubMed.
- T. J. Marks and J. R. Kolb, Chem. Rev., 1977, 77, 263–293 CrossRef CAS.
- L. J. Basile, J. R. Ferraro and D. Gronert, J. Inorg. Nucl. Chem., 1971, 33, 1047–1053 CrossRef CAS.
- L. M. Arnbjerg, D. B. Ravnsbæk, Y. Filinchuk, R. T. Vang, Y. Cerenius, F. Besenbacher, J. E. Jørgensen, H. J. Jakobsen and T. R. Jensen, Chem. Mater., 2009, 21, 5772–5782 CrossRef CAS.
- L. H. Brixner and E. P. Moore, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 1316 CrossRef.
- S. V. Mahajan, J. Hart, J. Hood, A. Everheart, M. L. Redigolo, D. S. Koktysh, E. A. Payzant and J. H. Dickerson, J. Rare Earths, 2008, 26, 131–135 CrossRef.
- S. J. Hwang, R. C. Bowman Jr, J. W. Reiter, J. Rijssenbeek, G. L. Soloveichik, J. C. Zhao, H. Kabbour and C. C. Ahn, J. Phys. Chem. C, 2008, 112, 3164–3169 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables including crystallographic data. Figures including IR, TG/DSC and in situ SR-PXD. This material is available free of charge via the internet. See DOI: 10.1039/c3ra44012e |
| This journal is © The Royal Society of Chemistry 2014 |