 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A pathway to diphosphorus from the dissociation of photoexcited tetraphosphorus†
Lee-Ping Wangb, Daniel Tofana, Jiahao Chena, Troy Van Voorhisa and Christopher C. Cummins*a
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, MA 02139, USA. E-mail: ccummins@mit.edu
bDepartment of Chemistry, Stanford University, Stanford, California, USA
First published on 25th September 2013
Abstract
We report a computational study of an energetically favorable pathway for the excited-state dissociation of a tetrahedral P4 molecule into two P2 molecules via the simultaneous breaking of four chemical bonds along a highly symmetric (D2d) reaction pathway. Along this pathway, a degeneracy occurs between the first excited state of P4 and the ground state of 2P2 at a lower total energy (ca. 4.7 eV) than the initial state, indicating that the initial photoexcitation provides sufficient energy for the dissociation without significant kinetic barriers. We also found that sequential dissociation of the four P–P bonds exhibits larger activation barriers thus making this a less viable dissociation pathway. Our computational investigation uncovers complicated photochemistry in elemental phosphorus, and suggests a likely mechanism for the environmentally friendly inclusion of phosphorus atoms into organic molecules.
1. Introduction
The idea of “cracking” P4 into two P2 molecules in order to take advantage of the high reactivity of the π bonds in the latter is attractive,1 but this process is not conveniently accessed thermally as very high temperatures (ca. 1100 K) are needed to break down the tetrahedral P4 molecule.2 On the other hand there have been indications since at least 1937 that UV irradiation of white phosphorus, a process that converts it into a red form, may initially bring about P2 generation.3 In one report on the P4 co-photolysis with metal carbonyl complexes, Dahl et al. mention that “P4 in solution photolyzes readily to P2 at ambient temperatures”.4 Co-photolysis of P4 with metal carbonyl complexes with formation of metal-phosphorus products has been reported in a few other cases,5 but only recently, Tofan and Cummins demonstrated the inclusion of diphosphorus moieties into organic molecules is afforded directly from the irradiation of mixtures of P4 and 1,3-dienes.6 The major organophosphorus products are cyclic compounds with a single P2 unit shared between the two fused six-membered rings. This reaction was proposed to occur through Diels–Alder additions of 1,3-diene molecules to photo-generated P2 units (Scheme 1).6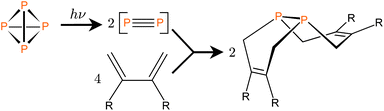 | ||
| Scheme 1 One-step synthesis of diphosphanes with the proposed involvement of P2 as a reaction intermediate.6 | ||
Here we report a theoretical investigation of the photolysis mechanism of P4. In this reaction, the starting point was an electronically excited state of a single P4 molecule and the endpoint was the ground state of two P2 molecules. We knew that this pathway must proceed through the dissociation of four chemical bonds and may involve a change in the electronic state. From a theoretical perspective, the problem is highly interesting in that the P4 is a small enough system to be treatable using highly accurate multi-reference quantum chemistry methods, yet the task of elucidating a low-energy pathway for photolysis involves a nontrivial exploration of the multidimensional potential energy surface (PES). In terms of experimental chemistry, an understanding of the P4 photolysis mechanism could lead to improved specificity and reaction yields for this type of photochemistry and increase the viability of this pathway as an environmentally friendly method for the incorporation of phosphorus atoms into organic molecules.7
In our explorations of the excited state PES, we found a surprising pathway for the direct dissociation of excited P4 into 2P2 that involves the simultaneous breaking of four P–P bonds. This pathway is easily visualized by placing the four phosphorus atoms on the corners of a cube such that no two atoms share a cube edge; the dissociation coordinate is equivalent to moving the top and bottom faces of the cube away from one another while contracting the P–P bonds in each diatomic subunit (Fig. 1), from 2.1994(3) Å in P4,7 to 1.8934 Å in P2.8 This result is counterintuitive because large reaction barriers would be expected for the simultaneous breaking of four chemical bonds. In this situation, we do not encounter any significant barriers because a state crossing exists along the dissociation coordinate in which the first excited state of P4 becomes degenerate with the ground state of 2P2. The state crossing occurs at a lower energy than the P4 excitation energy, so the photon energy is sufficient to carry out the dissociation.
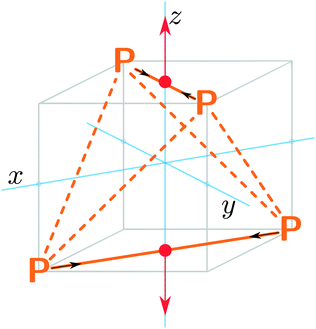 | ||
| Fig. 1 Dissociation of the P4 tetrahedron into two P2 units with simultaneous breaking of four P–P bonds occurs through a D2d-symmetric pathway. | ||
Although the precise mechanism of P4 → 2P2 photolysis contains interesting open questions, much is known about the molecules at the two endpoints of the reaction; here we briefly summarize the most important experimentally known properties and the corresponding theoretical calculations. Experimentally, the thermochemistry of the photolysis reaction is known. The reaction enthalpy for P4 → 2P2 is experimentally measured to be +53.8 ± 0.7 kcal mol−1;9 dissociation occurs spontaneously at 1100 K as the reaction becomes entropically driven.2 In terms of photochemistry, P4 is experimentally known to possess two low-lying excitations at 5.1 eV and 5.9 eV with more strongly absorbing manifolds of excitations above 7 eV.10–12 The low-lying excitations are interpreted as being HOMO–LUMO in character, although there are symmetry-imposed restrictions on the allowed transitions. Theoretically, the reaction enthalpy of P4 → 2P2 can be computed to within 1 kcal mol−1 of the experimental measurement; such an accurate estimation requires a treatment of dynamic electron correlation at the coupled cluster singles, doubles, and nonperturbative triples (CCSD(T)) level of theory using a nearly complete basis set.13 The UV absorption spectra of tetrahedral P4 and P2 have also been computed using the configuration interaction singles (CIS), random phase approximation (RPA) and equation-of-motion coupled cluster singles and doubles (EOM-CCSD) theoretical methods to within 0.5 eV (≈10 kcal mol−1) of the experimental measurement.14
2. Theory and computational methods
Our calculations are performed on gas-phase phosphorus clusters in the absence of a solvating environment. There are a number of theoretical studies on phosphorus clusters in the literature, but most of them have focused on equilibrium configurations of the ground electronic state. For example, Häser and Ahlrichs have investigated the equilibrium geometries and energies of many phosphorus clusters sometimes containing up to thirty atoms.14–16 These studies were carried out using methods including Hartree–Fock, density functional theory (DFT), second-order Moller–Plesset perturbation theory (MP2), and CCSD. We refer to these as single-reference electronic structure methods because they are based upon the assumption that the ground-state wavefunction is mostly described by a single electron configuration or Slater determinant, which is then used as a reference state for the calculations of electron correlation effects. Single-reference methods for the calculation of excited state energies include CIS, RPA, time-dependent density functional theory (TDDFT), and EOM-CCSD.The ground state of tetrahedral P4 and P2 are both closed-shell configurations with singlet spin multiplicity, and thus they are qualitatively well described by single Slater determinants. However, in general there is no reason to expect that the electronic state should stay the same throughout the entire dissociation pathway. Furthermore, the ground state of reaction intermediates may have multi-reference character (also known as static correlation); this means that the wavefunction cannot be described by a single Slater determinant. Multi-reference wavefunctions often appear in geometries where chemical bonds are broken and the system contains multiple radicals, and in these situations single-reference methods may be qualitatively incorrect. One would thus expect single-reference methods to perform well at the endpoints of the pathway but give unreasonable answers in the middle; this is indeed what we found in early exploratory calculations.
For this reason, we turn to complete active space self-consistent field (CASSCF), which is a method capable of describing static correlation; in CASSCF, the wavefunction is a linear combination of several Slater determinants, each of which represents a distinct electron configuration or orbital occupation. A CASSCF calculation is specified by choosing the number of active electrons and the number of active orbitals, and the wavefunction includes all configurations that can be constructed from placing the active electrons into the active orbitals.17 For example, in a CASSCF(2,3) calculation, there are two active electrons in three active orbitals, giving rise to six possible electron configurations, all of which are included in the wavefunction. The reliability of CASSCF calculations depends heavily on a good choice of the number of active electrons and the size of the active space.
In order to obtain quantitative accuracy, a calculation also needs to include dynamic correlation, which in the CASSCF picture corresponds to minuscule contributions to the energy from the electron configurations not considered in the wavefunction. Since the wavefunction does not contain sizable contributions from these configurations, they are not treated explicitly and instead are included using perturbation theory. Therefore, we expect that methods like CASSCF with second and third order corrections from Rayleigh–Schrödinger perturbation theory (RSPT2 and RSPT3) would provide quantitative accuracy for intermediate states on the pathway.
The CASSCF calculations took the symmetry of the geometries into account; orbitals were classified into the four irreducible representations of the D2 point group (A, B1, B2, B3). Along the entire dissociation pathway, the orbital occupations corresponding to the four representations was (9,7,7,7). The closed-shell orbitals were chosen to be (7,7,7,7) and the active space was chosen to be (3,2,2,2), because these frontier orbitals were well-separated from the occupied and virtual manifolds above and below for the entire dissociation process. The number of electronic states computed were (3,2,2,2) and state-averaging was performed within each representation. This makes the level of theory CASSCF(4,9)-RSPT3.
We verified our choice of active space with a CASSCF(12,13) calculation, where the active space includes all bonding electrons but is too large for applying accurate dynamic correlation treatments; here the closed-shell orbitals was (6,6,6,6) and the active space was (4,3,3,3). In both the (4,9) active space and the (12,13) active space, the CI vector describing the ground state and the orderings of the excited states stayed the same (ESI, Fig. S1 and S2†), providing verification that the (4,9) active space is a qualitatively correct description. We found that RSPT3 provided similar answers to RSPT2 but gave better agreement with the excitation energies in the tetrahedral geometry (ESI, Fig. S3†); this agreed with our intuition that RSPT3 should provide an improved treatment of dynamic correlation.
All calculations were performed with gas-phase structures using aug-cc-pVTZ basis sets and the MOLPRO software package.18
3. Results and discussion
Sequential dissociation coordinate.
In our initial explorations, we optimized the geometry of P4 in the first excited state. This caused the tetrahedral molecule to rearrange into a C2v-symmetric structure (Scheme 2, top middle) in which one of the P–P bonds was broken. From this geometry, an electronic relaxation to the ground state leads straight back to tetrahedral P4. The system is highly unlikely to follow minimum energy paths, however, since it possesses a large kinetic energy from the initial excitation; this motivated us to explore other potential pathways. We found another exothermic rearrangement leading to a D2d-symmetric minimum (Scheme 2, top right) with a low activation barrier of ≈0.5 eV in which one more P–P bond was broken. However, further bond dissociation involved much greater activation barriers of approximately ≈1.4 eV; here the system proceeds through a C2-symmetric transition state. Thus, a dissociation pathway passing through the molecular geometry critical points involving the sequential breaking of P–P bonds is possible, though it may require the system to possess large amounts of kinetic energy for crossing over the large activation barriers.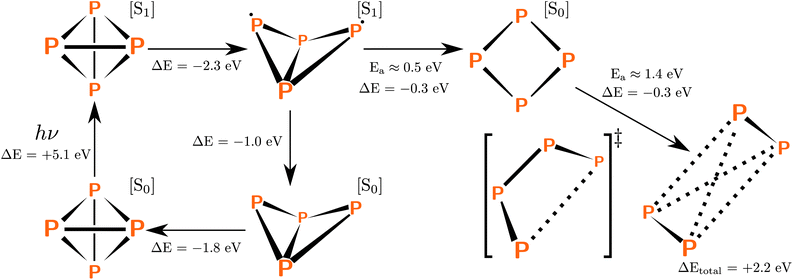 | ||
| Scheme 2 Molecular geometry critical points in the sequential dissociation pathway. The bottom left is the ground state of P4 and the far right is the ground state of 2P2. P4 enters the excited state after absorbing a photon, and a geometry optimization on the excited state surface leads to the C2v-symmetric geometry (top middle). From here, relaxation to the ground state leads back to tetrahedral P4, but rearrangement into a D2d-symmetric minimum is also possible (top right). Dissociation of the D2d-symmetric geometry into two 2P2 molecules occurs with a significant activation barrier of ≈1.4 eV. | ||
Direct dissociation coordinate.
With the results of the previous exploration in mind, we reasoned that excited-state P4 is likely to follow a highly ballistic trajectory without passing through the molecular geometry critical points.19 This motivated us to study a direct dissociation coordinate in which the opposite edges of the P4 tetrahedron were pulled apart linearly, involving the simultaneous breaking of four P–P bonds (Fig. 1). To our surprise we found that the energy along this coordinate was almost entirely downhill, with the exception of a very small barrier (0.1 eV) in the neighborhood of a state crossing.To generate the direct dissociation coordinate, we first optimized both the P4 and P2 structures at the MP2/aug-cc-pVTZ level. The dissociated geometry was built from placing two optimized P2 molecules 5 Å apart on the z-axis, with one P2 molecule placed symmetrically on the x-axis and the other on the y-axis. For comparison, the distance between the two P2 units within the optimized P4 molecule was found to be 1.56 Å. Fifty intermediate frames were generated by linearly interpolating from P4 to 2P2, corresponding to a 0.070 Å increase per frame in the distance between the two units.
Calculations were carried out for each point on the dissociation trajectory. At frame 0 (the tetrahedral geometry, z-axis separation = 1.56 Å), there are two threefold-degenerate excited states between 5 eV and 6 eV corresponding to the experimental optical excitation between the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO). As the dissociation proceeds, the degeneracy is lifted and the excited states in the B1 and B2 representations increase monotonically in energy (Fig. 2 depicts the A and B3 states, but not the B1 and B2 since these increase monotonically). The energy of the lowest B3 state decreases slightly until a minimum of 4.2 eV is reached at frame 5 (dimer separation = 1.90 Å), corresponding to an excimer-like geometry.20
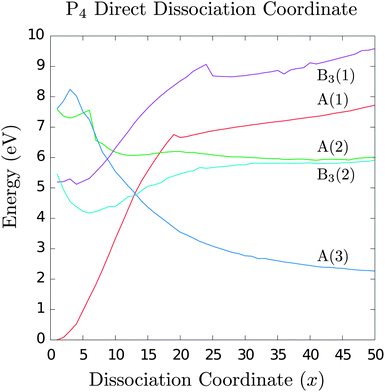 | ||
| Fig. 2 Energies of P4 electronic states along the direct dissociation coordinate. The experimental excitation corresponds to state B3(1) or B3(2). Note the appearance of a minimum in state B3(2) at x = 5, which we show later to be a saddle point on the many-dimensional potential surface. There is a near threefold degeneracy at x = 12 where the system may cross into the A(3) state; this corresponds to the ground state of two closed-shell P2 molecules. At dissociation, we recover the experimental P4 → 2P2 reaction enthalpy (compare asymptotic value of A(3) to starting value of A(1)). | ||
As we proceed along this coordinate, there is a degeneracy around 4.7 eV at frame 12 (z-axis separation = 2.50 Å) where the lowest B3 state intersects with the A(1) and A(3) states. It is unclear whether this is a true threefold degeneracy or three close intersections of twofold degeneracies. The most interesting result is that the A(3) electronic state corresponds to the ground state configuration of a P2 dimer, and the degeneracy occurs at a lower energy with respect to the initial excitation. Thus, this provides a possible pathway for the direct dissociation of photoexcited P4 into two P2 molecules. We note that at this degeneracy it is also possible to cross back to the ground state surface; this is expected to lead back to the tetrahedral geometry of ground state P4.
At the end of the trajectory, the geometry approaches that of two well-separated P2 molecules. The A(3) state becomes the ground state at the dissociation limit and is dominated by a single determinant of coefficient 0.94. The next highest states (A(1) and A(2)) become essentially degenerate excitations of single P2 dimers with a small exchange splitting. The excitation energy of approximately 3.8 eV is in good agreement with experimental absorption spectra of P2.21
The photochemistry along this pathway can be understood by examining the frontier orbitals (as shown in Fig. 3). At the tetrahedral P4 geometry, the HOMO shows bonding character across four edges of the tetrahedron and antibonding character across two edges, while the LUMO has complementary character with antibonding character across four edges and bonding character across two. Our intuition is that the direct dissociation coordinate involves an electronic state where two electrons from the HOMO are promoted into the LUMO, and this electronic state would correspond to the ground state of 2P2 at dissociation. This intuition is supported by examining the CI vector and MOs across the pathway.
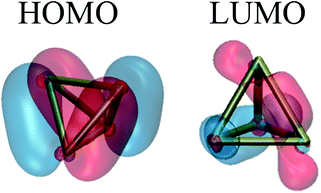 | ||
| Fig. 3 Highest occupied and lowest unoccupied molecular orbitals of P4. In the tetrahedral geometry, these orbitals are twofold and threefold degenerate, respectively. Note the bonding character of the HOMO and LUMO; the HOMO has bonding character across four edges of the tetrahedron and antibonding character across two edges, and vice versa for the LUMO. Qualitatively, P4 dissociation involves the transfer of two electrons from the HOMO into the LUMO, which breaks four P–P bonds and increases the bond order of the other two. | ||
Since the potential surface for P4 dissociation is a many-dimensional hypersurface, we decided to scan the potential surface in other dimensions orthogonal to the direct dissociation coordinate. We computed two-dimensional energy profiles of the ground state and lowest two excited states along the dissociation coordinate and two orthogonal deformation coordinates and plotted the results (Fig. 4 and 5). From the plots, we can see that the excimer-like geometry found in the one-dimensional profile is really a saddle point, and the true minima involve deformations in other coordinates (this is hardly surprising because our geometry optimizations led to the C2v-symmetric structure). However, the potential surface of the first excited state is smooth and mostly flat compared to the other two surfaces, lending some credence to our hypothesis that excited-state P4 may follow a highly ballistic trajectory.
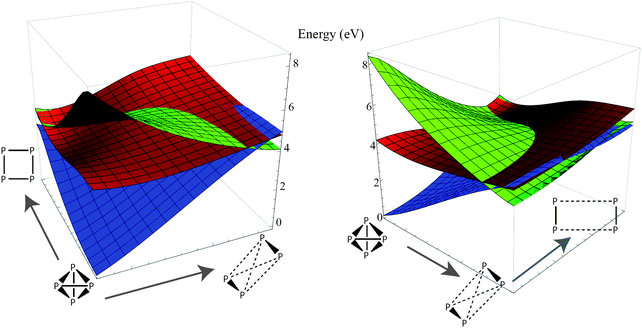 | ||
| Fig. 4 Two perspectives of a two-dimensional energy plot of the ground state (blue) and two lowest excited states (red, green). The coordinate to the right is the dissociation coordinate (3 ≤ x ≤ 15 in Fig. 2), and the orthogonal coordinate is a torsion about the dissociation vector (the range is roughly π/4). The coordinates in this plot have D2 symmetry. Note that the lowest excited state surface is relatively flat compared to the ground state, and there is a shallow minimum along the twisting coordinate. | ||
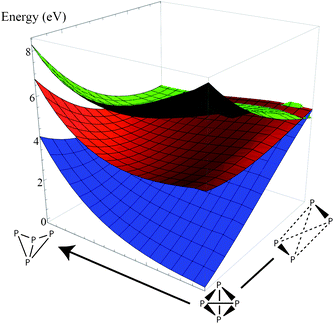 | ||
| Fig. 5 Two-dimensional energy plot of the ground state (blue) and two lowest excited states (red, green). The coordinate to the right is the dissociation coordinate (3 ≤ x ≤ 15 in Fig. 2), and the orthogonal coordinate is a torsion about the vector orthogonal to the dissociation vector and the P–P bond (the range is roughly π/4). The coordinates in this plot have C2 symmetry. Note that the lowest excited state surface is relatively flat compared to the ground state, and there is a shallow basin in the middle of the surface that is not visible in the one-dimensional dissociation plot. | ||
4. Conclusions
Our calculations of the excited-state potential surface of P4 show that it may be possible to dissociate P4 into 2P2 by the simultaneous breaking of four P–P bonds with the energy provided by UV irradiation. While there exist many pathways that lead back to the tetrahedral geometry, this does not rule out the possibility that a small population of molecules in the excited state may proceed along or near the direct dissociation pathway to generate small amounts of P2 in the reaction mixture. Qualitatively this is in agreement with the observations of Tofan and Cummins, in which a portion of the P4 reactant was recovered from the reaction mixture after twelve hours of UV illumination.6While our study suggests a possible pathway for the photolysis of P4 into 2P2, a firmer conclusion regarding the actual dissociation mechanism would necessitate further study using non-adiabatic molecular dynamics to explore the potential energy surfaces properly.22 Further studies of this nature may also shed light on other aspects of the photochemistry, such as the expected quantum yield for the photolytic reaction.
Acknowledgements
This work was funded by ENI S.p.A. as part of the Solar Frontiers Research Program. This material is based upon work supported by the National Science Foundation under Grant CHE-6923295 (T. V. V.) and CHE-1111357 (C. C. C.).References
- N. A. Piro, J. S. Figueroa, J. T. McKellar and C. C. Cummins, Science, 2006, 313, 1276–1279 CrossRef CAS PubMed.
- (a) D. P. Stevenson and D. M. Yost, J. Chem. Phys., 1941, 9, 403–408 CrossRef CAS; (b) H. Bock and H. Müller, Inorg. Chem., 1984, 23, 4365–4368 CrossRef CAS; (c) O. J. Scherer, Angew. Chem., Int. Ed., 2000, 39, 1029–1030 CrossRef CAS.
- G. Rathenau, Physica, 1937, 4, 503–514 CrossRef CAS.
- M. E. Barr, S. K. Smith, B. Spencer and L. F. Dahl, Organometallics, 1991, 10, 3983–3991 CrossRef CAS.
- (a) O. J. Scherer, J. Vondung and G. Wolmershuser, Angew. Chem., Int. Ed., 1989, 28, 1355–1357 CrossRef; (b) M. E. Barr and L. F. Dahl, Organometallics, 1991, 10, 3991–3996 CrossRef CAS; (c) O. J. Scherer, G. Berg and G. Wolmershäuser, Chem. Ber., 1996, 129, 53–58 CrossRef CAS; (d) M. Scheer and U. Becker, Chem. Ber., 1996, 129, 1307–1310 CrossRef CAS; (e) O. J. Scherer, T. Vömecke and G. Wolmershäuser, Eur. J. Inorg. Chem., 1999, 1999, 945–949 CrossRef.
- D. Tofan and C. C. Cummins, Angew. Chem., Int. Ed., 2010, 49, 7516–7518 CrossRef CAS PubMed.
- (a) B. M. Cossairt, C. C. Cummins, A. R. Head, D. L. Lichtenberger, R. J. F. Berger, S. A. Hayes, N. W. Mitzel and G. Wu, J. Am. Chem. Soc., 2010, 132, 8459–8465 CrossRef CAS PubMed; (b) M. Scheer, G. Balzs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed; (c) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed.
- G. Herzberg and K. Huber, Molecular spectra and molecular structure, Van Nostrand, 1979, pp. 518–521 Search PubMed.
- CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, 91st edn, 2010–2011, http://www.hbcpnetbase.com/ Search PubMed.
- R. R. Hart, M. B. Robin and N. A. Kuebler, J. Chem. Phys., 1965, 42, 3631–3638 CrossRef CAS.
- C. R. Brundle, H. Basch, N. A. Kuebler and M. B. Robin, Inorg. Chem., 1972, 11, 20–25 CrossRef CAS.
- M. E. Boyle, B. E. Williamson, P. N. Schatz, J. P. Marks and P. A. Snyder, Chem. Phys. Lett., 1986, 125, 349–354 CrossRef CAS.
- A. Karton and J. M. L. Martin, Mol. Phys., 2007, 105, 2499–2505 CrossRef CAS.
- M. Häser and O. Treutler, J. Chem. Phys., 1995, 102, 3703–3711 CrossRef.
- R. Ahlrichs, S. Brode and C. Ehrhardt, J. Am. Chem. Soc., 1985, 107, 7260–7264 CrossRef CAS.
- M. Häser, U. Schneider and R. Ahlrichs, J. Am. Chem. Soc., 1992, 114, 9551–9559 CrossRef.
- K. Andersson, P.-Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, MOLPRO, version 2010.1, a package of ab initio programs, 2010, http://www.molpro.net Search PubMed.
- B. K. Carpenter, Annu. Rev. Phys. Chem., 2005, 56, 57–89 CrossRef CAS PubMed.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CAS.
- K. P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure, Vol. IV in Constants of Diatomic Molecules, Von Nostrand, New York, 1979 Search PubMed.
- (a) H. R. Hudock, B. G. Levine, A. L. Thompson, H. Satzger, D. Townsend, N. Gador, S. Ullrich, A. Stolow and T. J. Martinez, J. Phys. Chem. A, 2007, 111, 8500–8508 CrossRef CAS PubMed; (b) J. C. Tully and R. K. Preston, J. Chem. Phys., 1971, 55, 562 CrossRef CAS; (c) M. Ben-Nun, J. Quenneville and T. J. Martinez, J. Phys. Chem. A, 2000, 104, 5161–5175 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: MOLPRO input file for the CASSCF calculations and molecular geometries along both dissociation coordinates. See DOI: 10.1039/c3ra43940b |
| This journal is © The Royal Society of Chemistry 2013 |